🤨 Mfuzz | 新瓶装旧酒,也是不一样的味道!~(经典的时间趋势分析包!~)
🤨 Mfuzz | 新瓶装旧酒,也是不一样的味道!~(经典的时间趋势分析包!~)

1写在前面
今天不太忙,但也不知道做什么,不知道是不是而立之年的悲哀!~😔
找不到方向,也不知道要干嘛,感觉每天都是:“哦,又活了一天。”😅
前面写了个TCseq,还有小伙伴想问经典的Mfuzz,其实这个包一开始是为microarray开发 。🤣
不过很多大paper里依然在用这个方法,大家一起看看吧。😷
2用到的包
rm(list = ls())
library(tidyverse)
library(Mfuzz)
3示例数据
data(yeast)
class(yeast)

这里的示例文件是个ExpressionSet格式,我们试试从头创建。😎
DEGs_exp_averp <- as.matrix(yeast@assayData$exprs)
boxplot(DEGs_exp_averp)
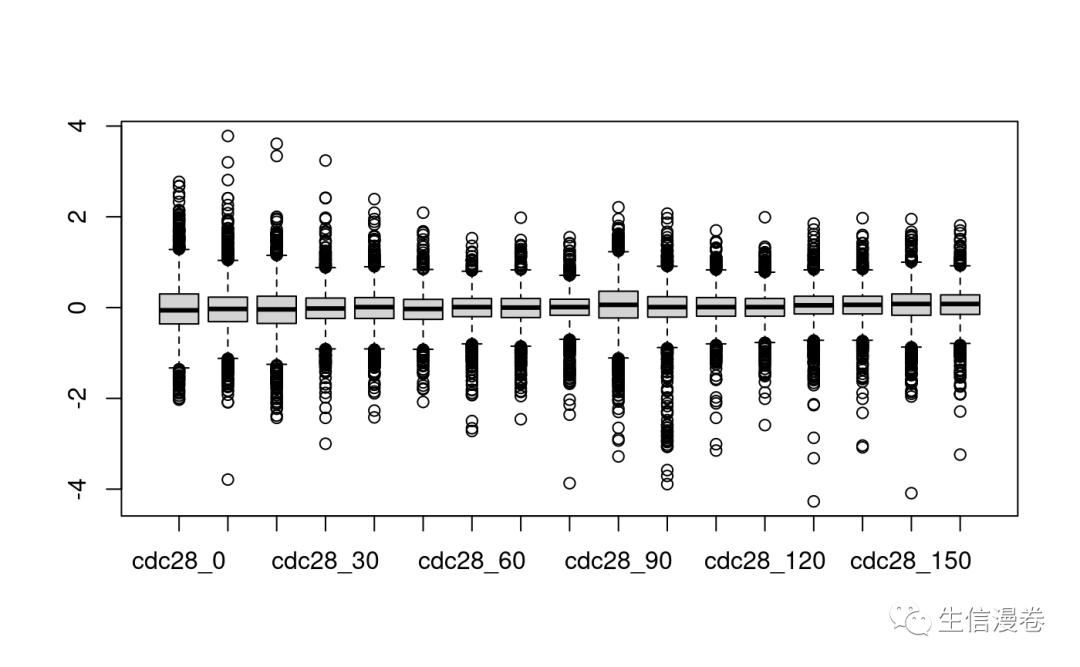
eset <- new('ExpressionSet',exprs = DEGs_exp_averp)
eset <- yeast
4预处理
4.1 缺失值的处理
首先我们过滤一下NA超过25%的基因。
eset <- Mfuzz::filter.NA(eset, thres = 0.25)

4.2 填补缺失值
方法很多哦,这里提供3种,mean, knn和wknn。😘
eset <- Mfuzz::fill.NA(eset, mode = 'mean')
#eset <- Mfuzz::fill.NA(eset,mode="knn")
#eset <- Mfuzz::fill.NA(eset,mode="wknn")
4.3 过滤基因
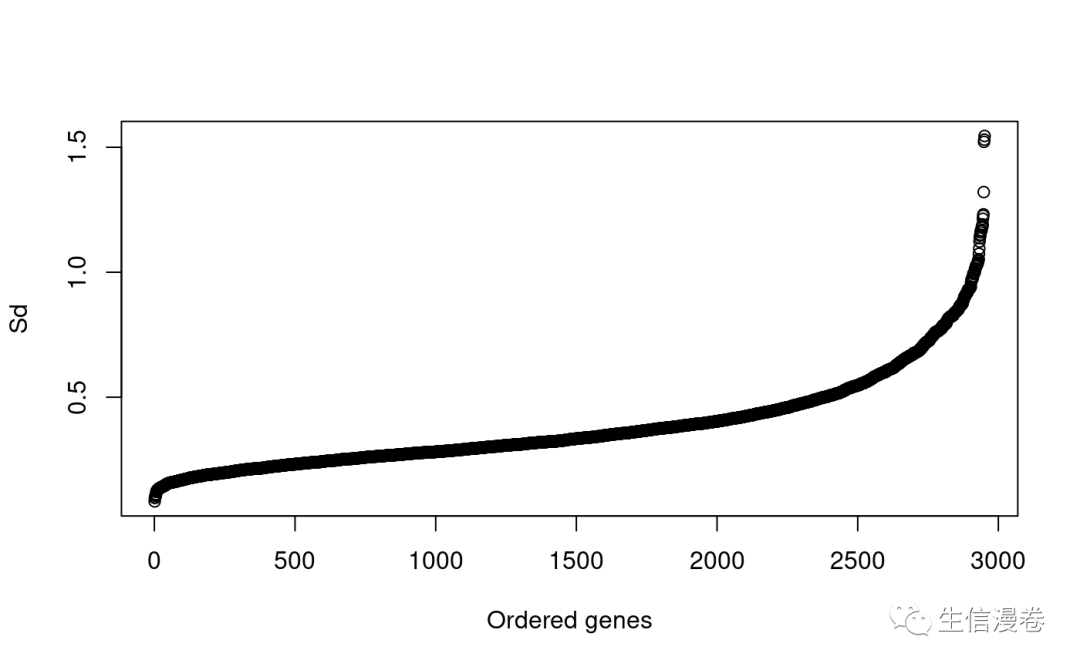
我们过滤一下低水平表达或仅显示表达微小变化的基因。🤓
tmp <- filter.std(eset,min.std=0)
4.4 标准化
Mfuzz默认输入的表达数据是经过归一化的。🤣
标准化不能代替哦。😘
eset.s <- standardise(tmp)
5聚类
c <- 16
m <- mestimate(eset.s)
set.seed(123)
cl <- mfuzz(eset.s, c = c, m = m)
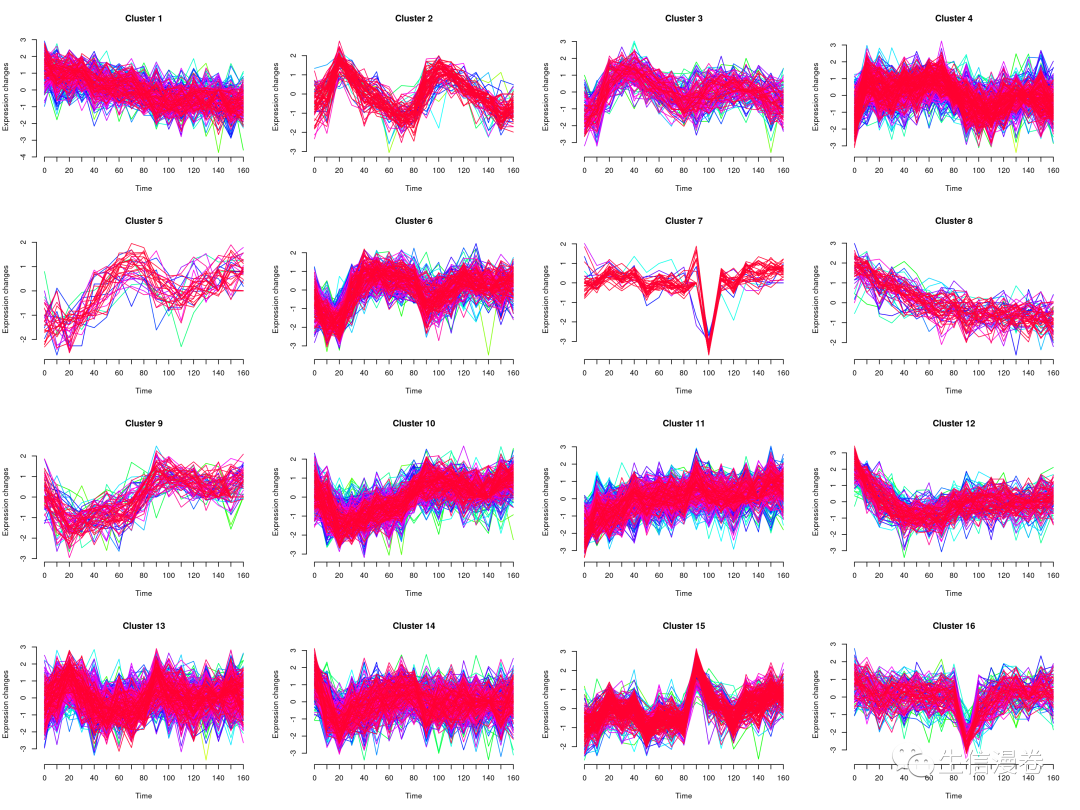
6可视化
mfuzz.plot(eset.s, cl=cl, mfrow=c(4,4), new.window = F,time.labels = seq(0, 160, 10))
7提取聚类结果
查看每个cluster中的基因个数。🤪
cl$size

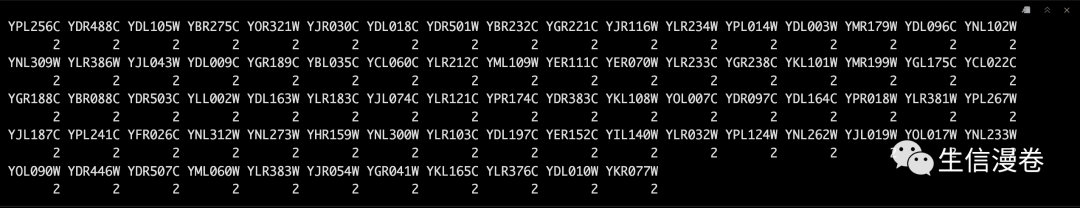
提取某个cluster下的基因试试呢,比如2。🧐
cl$cluster[cl$cluster == 2]
查看基因和cluster之间的membership。😎
非常有用哦!~🤩
head(cl$membership)
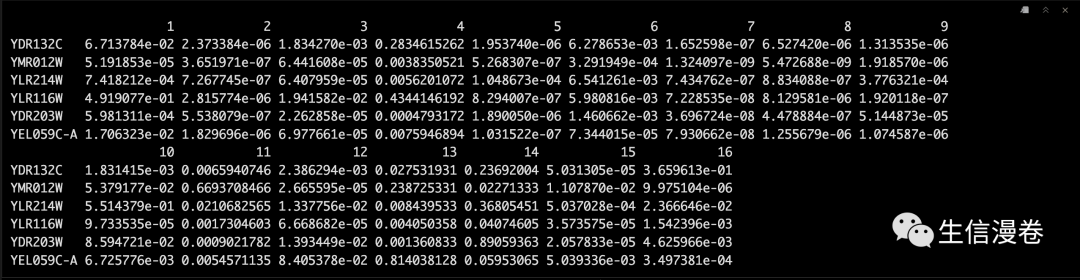
最后就输出结果吧。😅
output <- data.frame(eset.s@assayData$exprs)
output$cluster <- cl$cluster
#write.csv(as.data.frame(output),file = "./Mfuzz.csv",colNames = T,rowNames=F)

最后祝大家早日不卷!~
点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 往期精彩
📍 🤩 LASSO | 不来看看怎么美化你的LASSO结果吗!? 📍 🤣 chatPDF | 别再自己读文献了!让chatGPT来帮你读吧!~ 📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~ 📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码! 📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!? 📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案) 📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程 📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~ 📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~ 📍 🧐 rms | 批量完成你的线性回归 📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图 📍 🤠 Network | 高颜值动态网络可视化工具 📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图 📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程) 📍 ......