🤩 Monocle 3 | 太牛了!单细胞必学R包!~(七)(分析单细胞轨迹中的分支)
🤩 Monocle 3 | 太牛了!单细胞必学R包!~(七)(分析单细胞轨迹中的分支)

生信漫卷
发布于 2023-12-19 16:15:53
发布于 2023-12-19 16:15:53
1写在前面
今天是大雪⛄️,完全没有想到雪能这么大,不知道明天值班要打多少石膏哦。🤒
有时候你感兴趣的只是单细胞轨迹中的一个分支,A → B。
这个时候就可以用到monocle3中一个非常实用的功能了。😘
我们一起来看看怎么操作吧。😬
2用到的包
rm(list = ls())
library(tidyverse)
library(monocle3)
3示例数据
今天做一做如何分析单细胞轨迹中的分支。😏
我们还是把之前的embryo数据加载进来用一下。🤩
expression_matrix <- readRDS("./packer_embryo_expression.rds")
cell_metadata <- readRDS("./packer_embryo_colData.rds")
gene_annotation <- readRDS("./packer_embryo_rowData.rds")
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
cds
4预处理
和之前一模一样,就不做过多介绍了。😂
cds <- preprocess_cds(cds, num_dim = 50)
cds <- align_cds(cds, alignment_group = "batch",
residual_model_formula_str = "~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading")
cds <- reduce_dimension(cds)
cds <- cluster_cells(cds)
cds <- learn_graph(cds)
cds <- order_cells(cds)
5选择轨迹
分析围绕轨迹分支节点调节的基因可以深入了解控制细胞命运决定的基因。🧬
做法非常简单choose_cells()即可,交互式。😘
cds_subset <- choose_cells(cds)
6识别相关基因集
然后调用graph_test()子集,识别出具有制定表达模式的基因,这些基因只落在你选择的轨迹区域内哦。😲
subset_pr_test_res <- graph_test(cds_subset, neighbor_graph="principal_graph", cores=4)
pr_deg_ids <- row.names(subset(subset_pr_test_res, q_value < 0.05))
7寻找模块
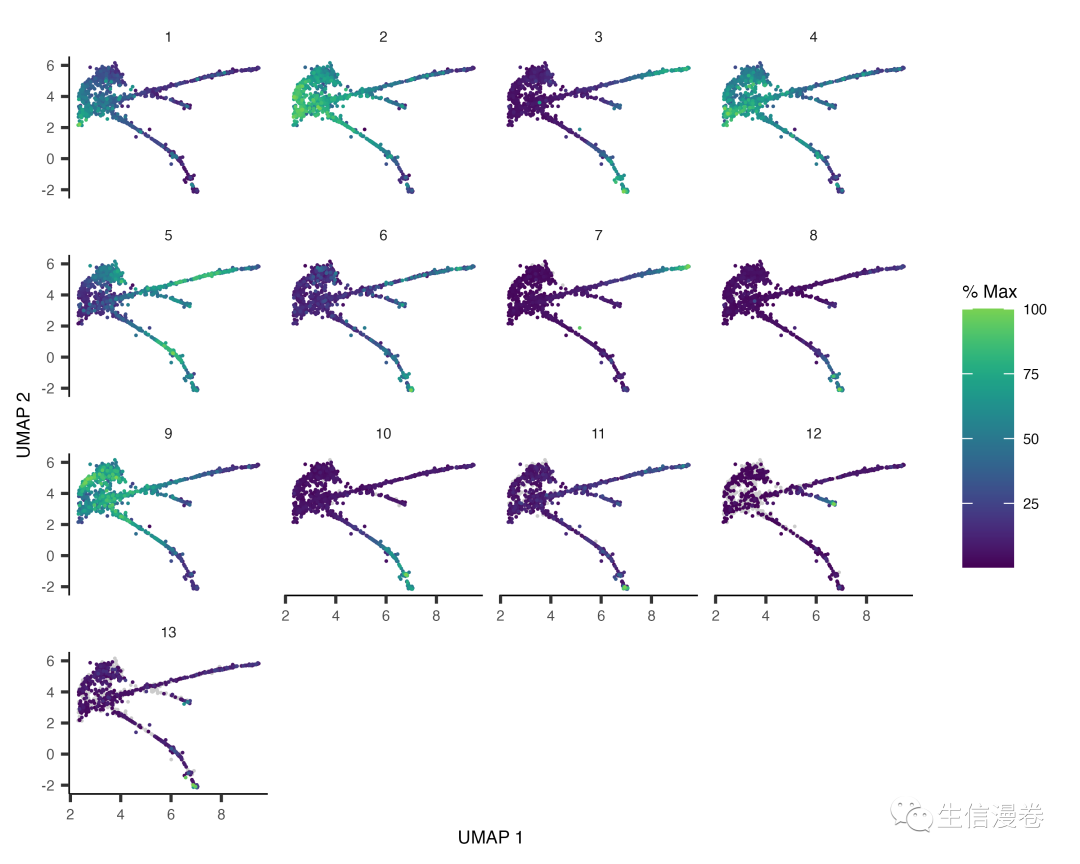
将这些基因分组到模块中可以揭示命运特异性基因或那些在分支节点之前或之后立即激活的基因:
gene_module_df <- find_gene_modules(cds_subset[pr_deg_ids,], resolution=0.001)
接着我们根据模块在轨迹上的相似性(hclust)来组织模块,以便更容易看到哪些模块先于其他模块出现:👇
agg_mat <- aggregate_gene_expression(cds_subset, gene_module_df)
module_dendro <- hclust(dist(agg_mat))
gene_module_df$module <- factor(gene_module_df$module,
levels = row.names(agg_mat)[module_dendro$order])
plot_cells(cds_subset,
genes=gene_module_df,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)
最后祝大家早日不卷!~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-12-18,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录