scBayes——整合bulk DNA与single-cell RNA进行肿瘤亚克隆表达研究的工具
scBayes——整合bulk DNA与single-cell RNA进行肿瘤亚克隆表达研究的工具

生信技能树
发布于 2024-03-25 15:40:32
发布于 2024-03-25 15:40:32
文章:Qiao Y, Huang X, Moos PJ, Ahmann JM, Pomicter AD, Deininger MW, Byrd JC, Woyach JA, Stephens DM, Marth GT. (2024) A Bayesian framework to study tumor subclone-specific expression by combining bulk DNA and single-cell RNA sequencing data. Genome Res
基因遗传与基因表达差异层面是许多肿瘤的基本标志,这种异质性能够不断发展从而对治疗产生耐药性。目前最常用的方法是bulk tumor/normal whole-genome or whole-exome sequencing (WGS, WES) 或者 scRNA-seq,但缺少将二者整合起来同时分析基因组肿瘤亚克隆性与转录组异质性的工具。
犹他大学的研究人员开发了scBayes,基于贝叶斯框架,使用bulk DNA测序数据推断的肿瘤亚克隆结构,通过单细胞基因表达确定细胞的亚克隆身份。该工具将代表相同遗传背景的肿瘤亚克隆的细胞组合在一起,从而比较不同亚克隆之间的基因表达,或研究同一亚克隆内基因表达随时间(比如进展、治疗反应或复发)或空间(在多个转移部位和器官)的变化。
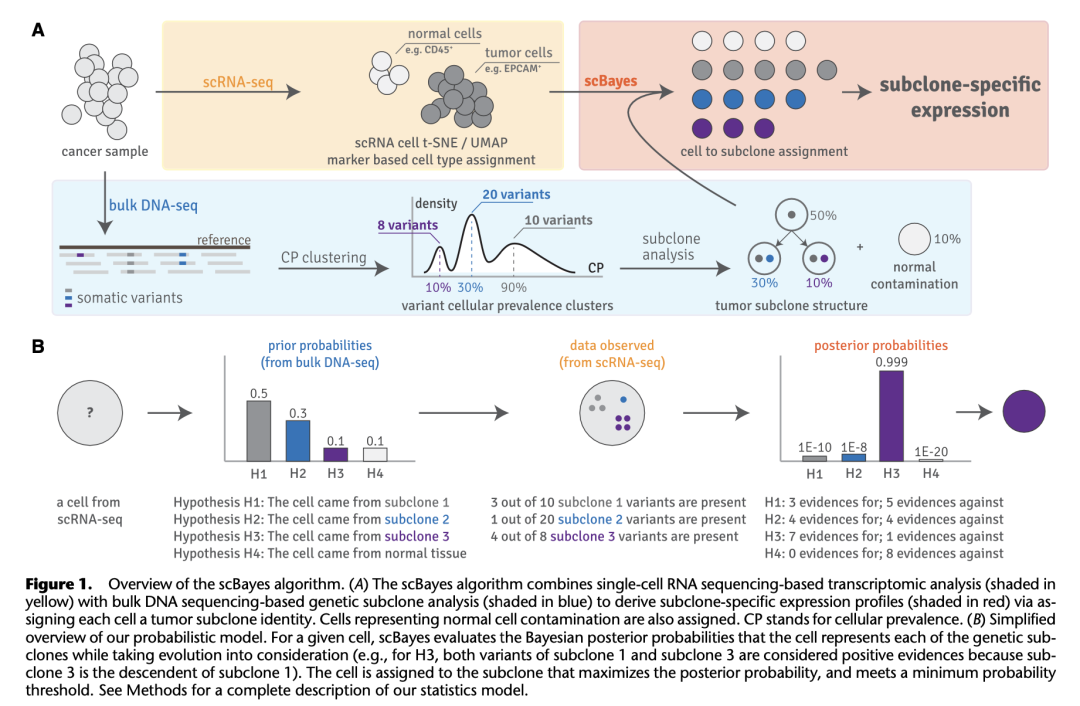
该方法适用于Smart-seq和10x,代码在:https://gitlab.com/yiq/scbayes
本文参与 腾讯云自媒体分享计划,分享自微信公众号。
原始发表:2024-03-21,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读