GEO多数据集联合分析-文献复现
原创
文献题目:基于生物信息学的新型铁死亡基因生物标志物和免疫浸润谱在糖尿病肾病中的应用Huang, Y., & Yuan, X. (2024). Novel ferroptosis gene biomarkers and immune infiltration profiles in diabetic kidney disease via bioinformatics. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 38(2), e23421. https://doi.org/10.1096/fj.202301357RR. IF: 4.8 Q1
本文下载GSE30122和GSE47185数据集表达数据,去除批次效应,整合到合并的数据集中,然后进行功能富集分析。然后筛选潜在的差异表达基因。鉴定铁死亡相关差异表达基因,然后进行基因本体分析。构建蛋白质-蛋白质相互作用网络并筛选枢纽基因。使用适当的算法评估数据集中的免疫细胞浸润状态。使用共识聚类分析构建免疫特征亚型。
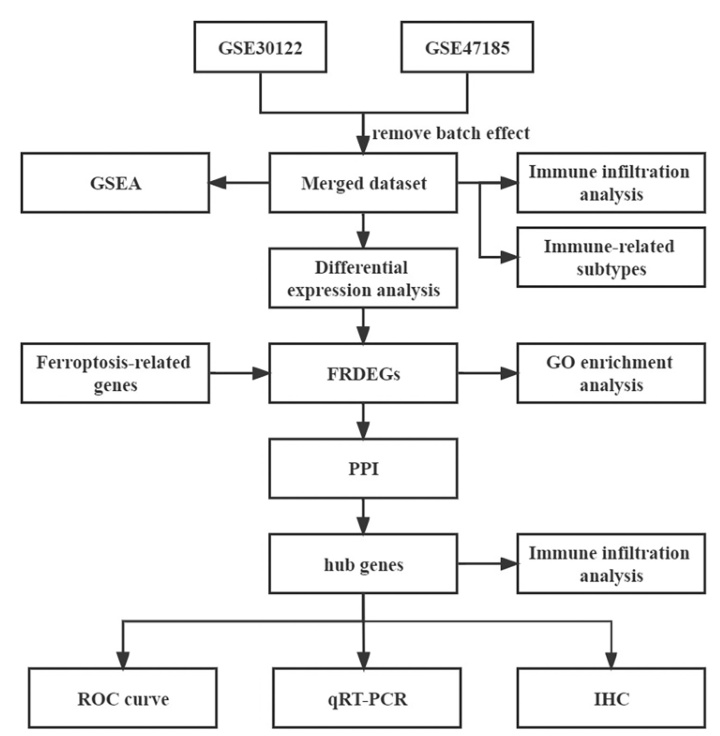
技术路线图
1. 找数据集及下载数据
GSE30122
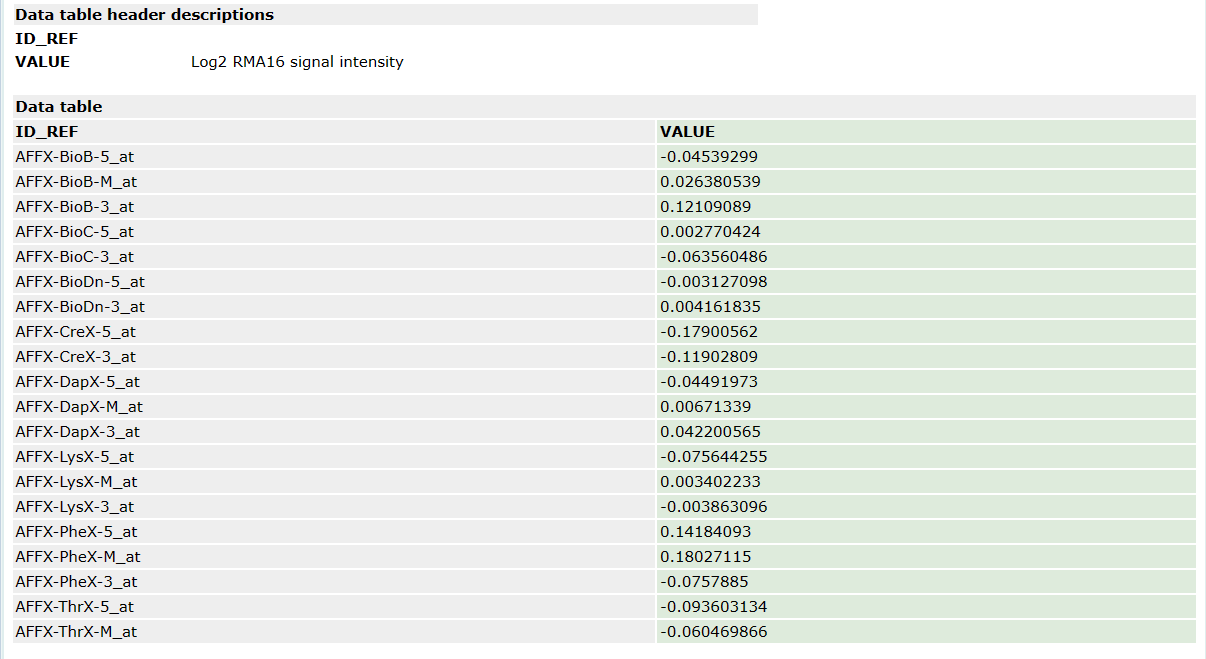
查看GSE30122的表达矩阵
可见数据有异常,有很多负值。那则需要下载原始数据
GSE30122的原始数据
芯片原始数据处理
rm(list = ls())
library(GEOquery)
library(stringr)
#芯片原始数据处理
BiocManager::install(c( 'oligo' ),ask = F,update = F)
library(oligo)
BiocManager::install(c( 'pd.hg.u133.plus.2' ),ask = F,update = F)
library(pd.hg.u133.plus.2)
dir='D:\\ferroptosis_diabetic_kidney\\GSE30122_RAW'
od=getwd()
setwd(dir)
celFiles <- list.celfiles(listGzipped = T)
celFiles
affyRaw <- read.celfiles( celFiles )
setwd(od)
eset <- rma(affyRaw)
eset
colnames(eset)
#列名需要修改以获取临床信息
colnames(eset) <- str_sub(colnames(eset),end = 9)
exp1 <- exprs(eset)
dim(exp1)
exp1[1:4,1:4]
#表达矩阵已取log, 不用再取log
boxplot(exp1, las = 2)
#提取临床信息
eSet1 <- getGEO("GSE30122",
destdir = '.',
getGPL = F)
pd1 <- pData(eSet1[[1]])
if(!identical(rownames(pd1),colnames(exp1))) exp1 = exp1[,match(rownames(pd1),colnames(exp1))]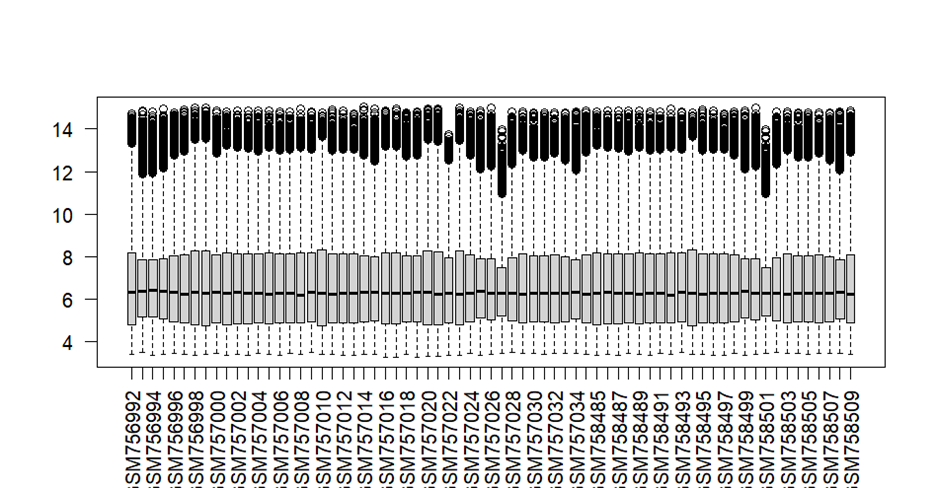
表达矩阵已取log, 不用再取log
第二个数据集下载及数据处理
#数据下载
eSet2 <- getGEO("GSE47185",
destdir = '.',
getGPL = F)
#(1)提取表达矩阵exp
exp2 <- exprs(eSet2[[1]])
exp2[1:4,1:4]
dim(exp2)
boxplot(exp2)
#(2)提取临床信息
pd2 <- pData(eSet2[[1]])
#GSE47185在文中作者提取了14个DN样本
str_detect(pd2$title,"Diabetic Nephropathy")
pd2 <- pd2[str_detect(pd2$title,"Diabetic Nephropathy"), ]
#只保留14个样本的表达矩阵
if(!identical(rownames(pd2),colnames(exp2))) exp2 = exp2[,match(rownames(pd2),colnames(exp2))]
dim(exp2)
boxplot(exp2,las = 2)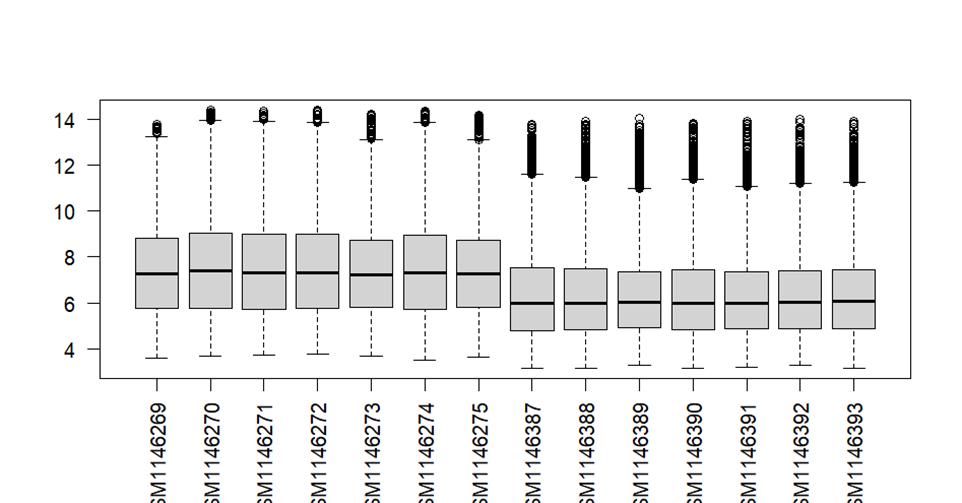
表达矩阵也有问题样本
# 样本矫正
library(limma)
exp2=normalizeBetweenArrays(exp2)
boxplot(exp2,las=2)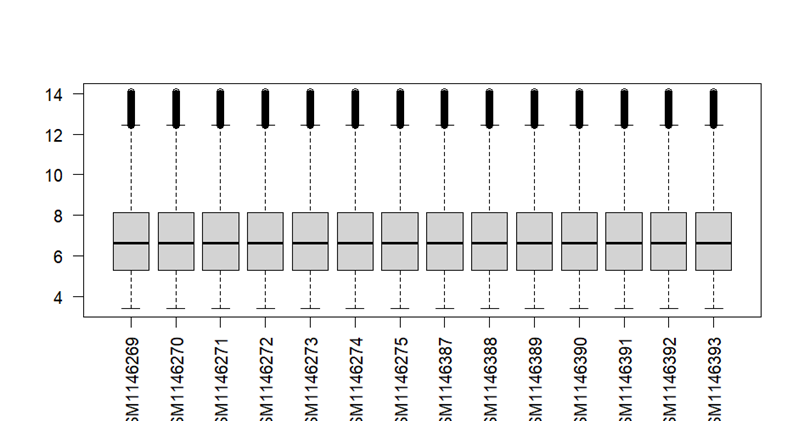
2.分组及探针获取
#(3)提取芯片平台编号
gpl1 <- eSet1[[1]]@annotation
gpl2 <- eSet2[[1]]@annotation
#(4)探针注释的获取
#由于来源于不同平台的芯片数据导致表达矩阵行名不一样,
#所以先分别注释成gene symbol,再合并表达矩阵,去除批次效应
library(tinyarray)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("hgu133a2.db")
find_anno(gpl1)
library(hgu133a2.db);ids <- toTable(hgu133a2SYMBOL)
ids1 <- ids
#为exp数据框添加几列
exp1 <- as.data.frame(exp1)
#1.加probe_id列,把行名变成一列
library(dplyr)
exp1 = mutate(exp1,probe_id = rownames(exp1))
#2.加上探针注释
ids1 = distinct(ids1,symbol,.keep_all = T)
#其他去重方式在zz.去重方式.R
exp1 = inner_join(exp1,ids1,by="probe_id")
nrow(exp1) #如果行数为0就是你找的探针注释是错的。
rownames(exp1) <- exp1[,71]GSE47185数据集注释没有R包,下载GEO注释文件进行注释
#GSE47185数据集注释
find_anno(gpl2)
#没有相应的R包,自行从GEO网页界面下载平台注释文件并打开编辑保留所需信息
ids2 <- read.table("GPL11670_family.soft", header = T, sep = "\t")
#为exp数据框添加几列
exp2 <- as.data.frame(exp2)
#1.加probe_id列,把行名变成一列
library(dplyr)
exp2 = mutate(exp2,GeneID = rownames(exp2))
#2.加上探针注释
ids2 = distinct(ids2,ORF,.keep_all = T)
ids2 = ids2[, 2:3]
#其他去重方式在zz.去重方式.R
exp2 = inner_join(exp2,ids2,by="GeneID")
ids2$GeneID <- as.character (ids2$GeneID)
exp2 = inner_join(exp2,ids2,by="GeneID")
nrow(exp2) #如果行数为0就是你找的探针注释是错的。
rownames(exp2) <- exp2[,16]3. 数据整合并去除批次效应
#(4)合并表达矩阵
table(rownames(exp1) %in% rownames(exp2))
length(intersect(rownames(exp1),rownames(exp2)))
exp1 <- exp1[intersect(rownames(exp1),rownames(exp2)),]
exp2 <- exp2[intersect(rownames(exp1),rownames(exp2)),]
exp1 <- exp1[,1:69]
exp2 <- exp2[,1:14]
exp = cbind(exp1,exp2)
boxplot(exp)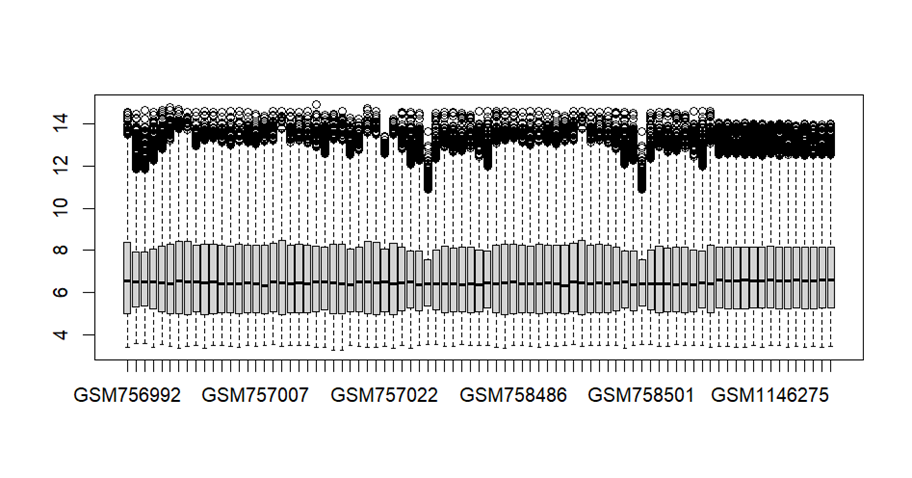
Group1 = ifelse(str_detect(pd1$title,"Control"),"Normal", "DN")
Group2 = ifelse(str_detect(pd2$characteristics_ch1.1,"Diabetic Nephropathy"),"DN","Normal")
Group = c(Group1,Group2)
table(Group)
Group = factor(Group,levels = c("Normal","DN"))
save(Group,exp,file = "exp.Rdata")
dimnames <- list(rownames(exp),colnames(exp))
data <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
dim(data)
class(data)
group_list <- data.frame(
sample = colnames(exp), c(rep("GSE30122",69),rep("GSE47185",14)))
rownames(group_list) <- group_list$sample
colnames(group_list)[2] <- "dataset"
group <- factor(group_list$dataset)
View(group_list)
library(FactoMineR)##没有请先安装
library(factoextra)
pdf(file = "PCA_before1.pdf",width = 7,height = 6)
pre.pca <- PCA(t(exp),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = group,
addEllipses = TRUE,
legend.title="Group"
)
dev.off()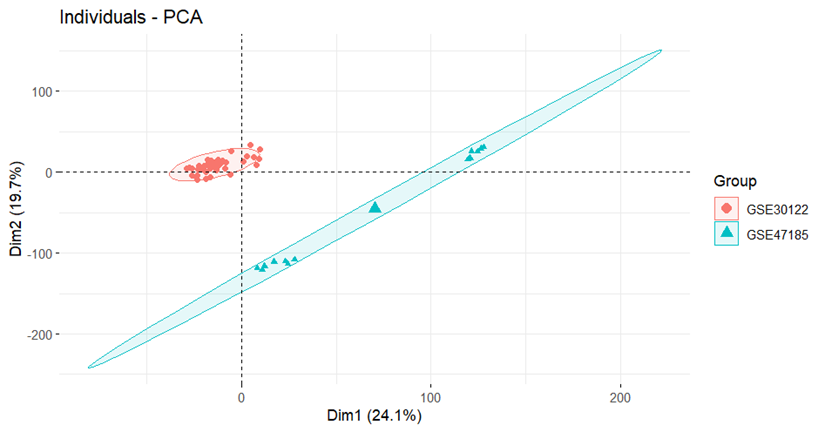
#-------------------进行去批次------------------#
#### 使用sva包计算批次效应
library(sva)
exp_combat <- ComBat(exp, batch = group_list$dataset) # batch为批次信息
boxplot(exp_combat)
# 查看去除批次效应的结果如何
library(tinyarray)
draw_pca(exp = exp, group_list = group)
#做校正的PCA分析
pdf(file = "9_PCA_after.pdf",width = 7,height = 6)
pre.pca <- PCA(t(exp_combat),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = group,
addEllipses = TRUE,
legend.title="Group")
dev.off()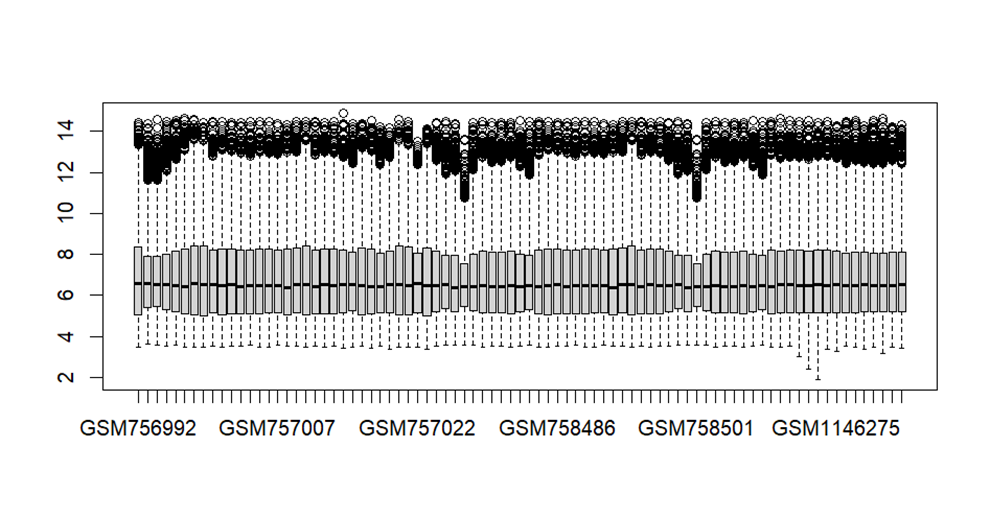
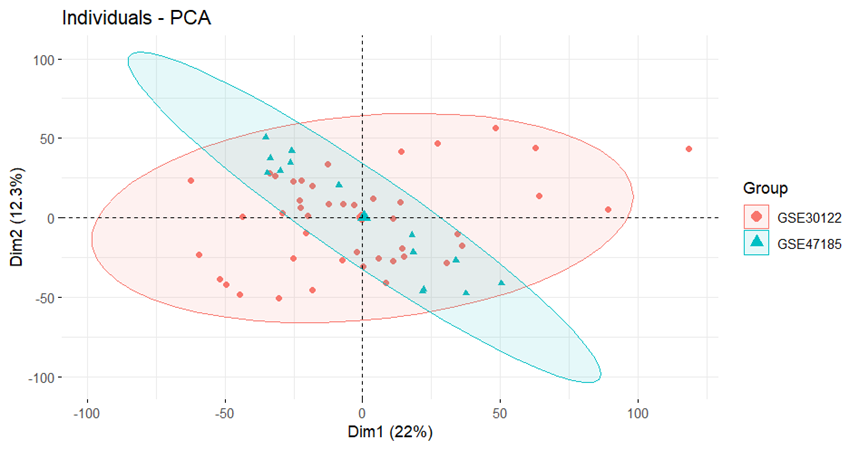
4.差异分析
因跟文中的样本表达矩阵处理不一样,故使用文中的差异基因的条件得到结果不一致
此处使用常用的筛选条件
#差异分析
library(limma)
design = model.matrix(~Group)
fit = lmFit(exp,design)
fit = eBayes(fit)
deg = topTable(fit,coef = 2,number = Inf)
#3.加change列,标记上下调基因
logFC_t = 1.0
p_t = 0.05
#思考,如何使用padj而非p值
k1 = (deg$P.Value < p_t)&(deg$logFC < -logFC_t)
k2 = (deg$P.Value < p_t)&(deg$logFC > logFC_t)
deg = mutate(deg,change = ifelse(k1,"down",ifelse(k2,"up","stable")))
table(deg$change)
#火山图
library(ggplot2)
ggplot(data = deg, aes(x = logFC, y = -log10(P.Value))) +
geom_point(alpha=0.4, size=3.5, aes(color=change)) +
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept=c(-logFC_t,logFC_t),lty=4,col="black",linewidth=0.8) +
geom_hline(yintercept = -log10(p_t),lty=4,col="black",linewidth=0.8) +
theme_bw()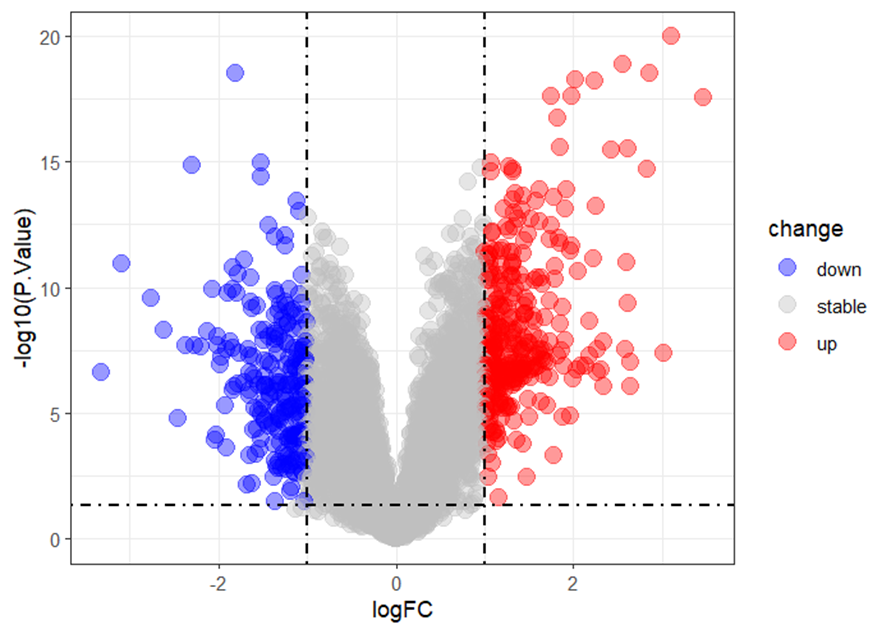
5.与铁死亡基因取交集,获得FRDEGs
#与ferroptosis- related genes (FRGs)取交集画韦恩图
#下载并读取文中FRGs文件,文中附件2即是。需整理成一列的TXT文件
deg = mutate(deg,symbol = rownames(deg))
DEG = deg$symbol[deg$change !="stable"]
FRG <- read.table("FRG.txt")
FRG <- FRG[,1]
library (VennDiagram)
venn.diagram(x= list(DEG = DEG,FRG = FRG),
filename = "pic.png",
height = 450, width = 450,
resolution =300,
imagetype="png",
col="transparent",
fill=c('#0099CC','#FF6666'),
alpha = 0.50,
cex=0.45,
cat.cex=0.45,)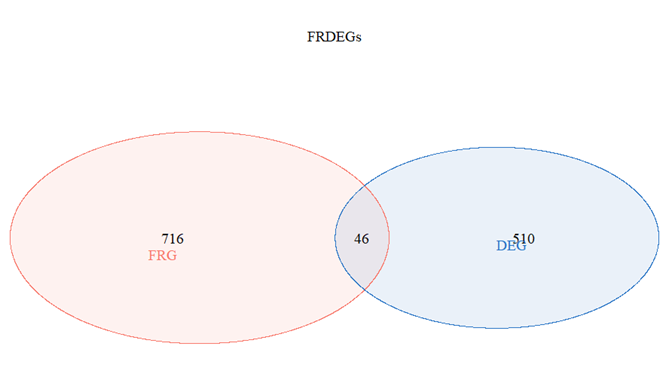
# FRDEGs热图----
# 表达矩阵行名替换为基因名
FRDEGs <- intersect(DEG,FRG)
exp = exp[deg$probe_id,]
rownames(exp) = deg$symbol
diff_gene = deg$symbol[deg$change !="stable"]
n = exp[FRDEGs,]
library(pheatmap)
annotation_col = data.frame(Group)
rownames(annotation_col) = colnames(n)
pheatmap(n,show_colnames =F,
show_rownames = T,
scale = "row",
cluster_cols = F,
annotation_col=annotation_col,
breaks = seq(-3,3,length.out = 100)
) 
6. 富集分析
#4.对FRDEGs加ENTREZID列,用于富集分析(symbol转entrezid,然后inner_join)
FRDEG_gene <- deg[FRDEGs,]
library(clusterProfiler)
library(org.Hs.eg.db)
s2e = bitr(FRDEG_gene$symbol,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)#人类,注意物种
#一部分基因没匹配上是正常的。<30%的失败都没事。
#其他物种http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
nrow(FRDEG_gene)
FRDEG_gene = inner_join(FRDEG_gene,s2e,by=c("symbol"="SYMBOL"))
#多了几行少了几行都正常,SYMBOL与ENTREZID不是一对一的。
nrow(FRDEG_gene)
save(exp,Group,FRDEG_gene,logFC_t,p_t,file = "step4output.Rdata")
rm(list = ls())
load(file = 'step4output.Rdata')
library(clusterProfiler)
library(ggthemes)
library(org.Hs.eg.db)
library(dplyr)
library(ggplot2)
library(stringr)
library(enrichplot)
#(1)输入数据
gene_diff = FRDEG_gene$ENTREZID
#(2)富集
ekk <- enrichKEGG(gene = gene_diff,organism = 'hsa')
ekk <- setReadable(ekk,OrgDb = org.Hs.eg.db,keyType = "ENTREZID")
ego <- enrichGO(gene = gene_diff,OrgDb= org.Hs.eg.db,
ont = "ALL",readable = TRUE)
#setReadable和readable = TRUE都是把富集结果表格里的基因名称转为symbol
class(ekk)
#(3)可视化
dotplot(ego, split = "ONTOLOGY") +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y")
dotplot(ekk)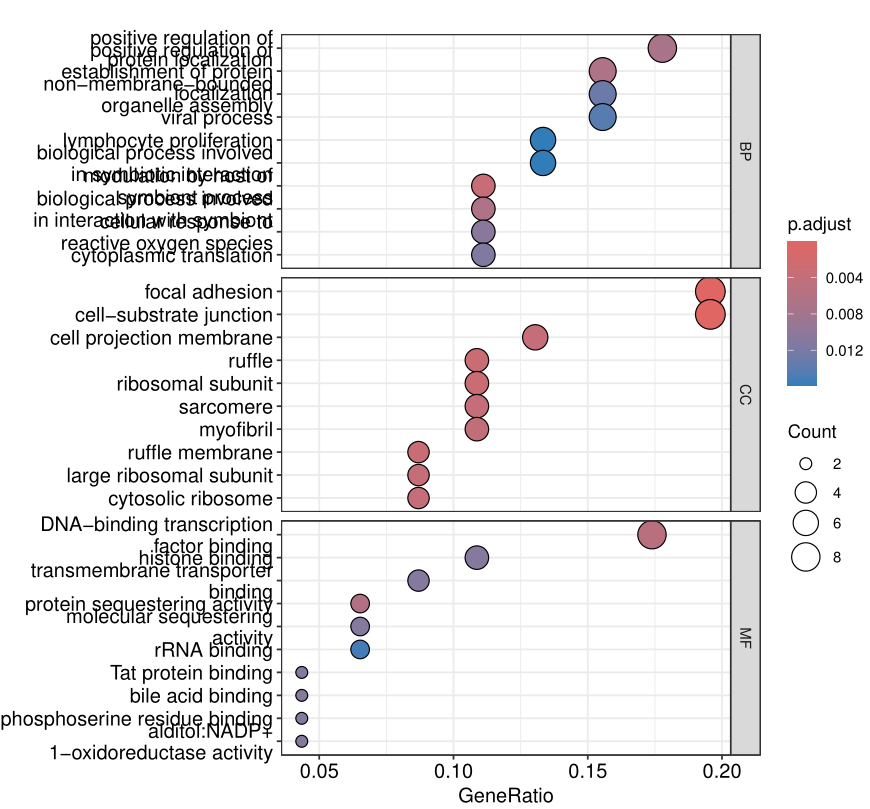
GO富集
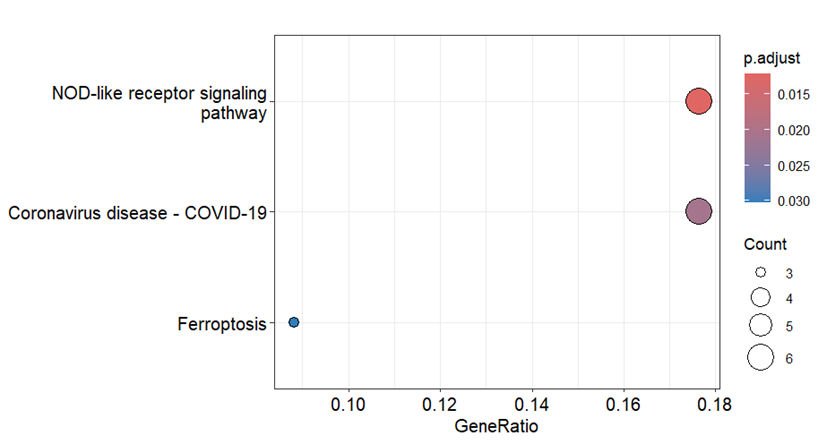
KEGG富集
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录