单细胞数据标准化及高变基因鉴定

单细胞下游分析流程及函数
- 数据读取:使用对应的函数读取相应格式的单细胞数据
- 创建seurat对象:CreateSeuratObject()
- 数据质控:基于nFeature_RNA及percent.mt等进行过滤
- 数据标准化:NormalizeData()
- 选择高变基因:FindVariableFeatures()
- 数据缩放:ScaleData()
- PCA降维:RunPCA()
- 细胞聚类分群:FindNeighbors()和FindClusters()
- TSNE/UMAP可视化:RunTSNE()及RunUMAP()
- 计算所有簇的Marker基因并可视化:FindAllMarkers()
- 细胞亚群命名:RenameIdents()或者SetIdent()
咱们的#单细胞常见图表系列推文,首先会按照单细胞下游分析基本流程整理里面的常见的图表,以及分析的过程
前几篇推文都是基于数据质控的,分享了如何使用下游数据查看细胞鉴定曲线,以及基于nFeature_RNA及percent.mt等进行过滤
那这篇推文我们一起来学习一下,数据的标准化以及选择高变基因可视化
数据标准化
在单细胞测序中,由于每个细胞的起始转录分子量很低(starting material),极低的转录分子量导致转录本的捕获和扩增效率存在技术差异,因此在文库构建中很难保证单细胞数据的高度一致性。从而造成多个样本的测序数据会因为文库测序覆盖率的不同而引入系统差异。
对数据进行过滤,删除掉一些不需要的数据后,为了排除技术误差,更好地呈现基因水平的差异,所以需要对数据进行标准化
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize",
scale.factor = 10000)
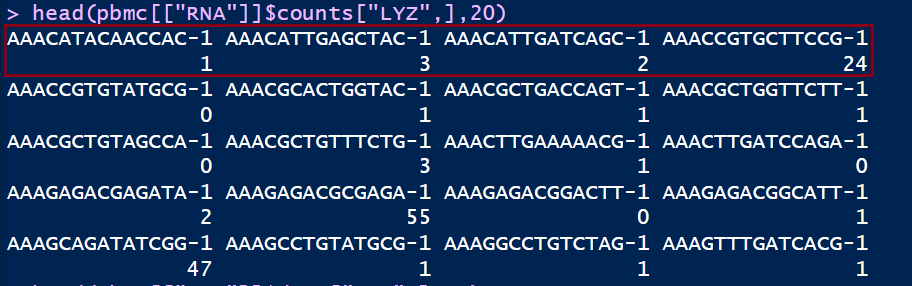
使用到的是NormalizeData()函数,一般采用全局缩放归一化方法“LogNormalize”(也有“CLR”和“RC”),该方法通过总表达量对每个单元格的特征表达值进行归一化,使用每个细胞的某个gene_counts除以该细胞的总的counts(nCount_RNA),再乘以一个比例因子(默认为10,000),并对结果进行自然对数变换。
#计算方法
log(10000*head(pbmc[["RNA"]]$counts["LYZ",])/head(pbmc$nCount_RNA)+1)
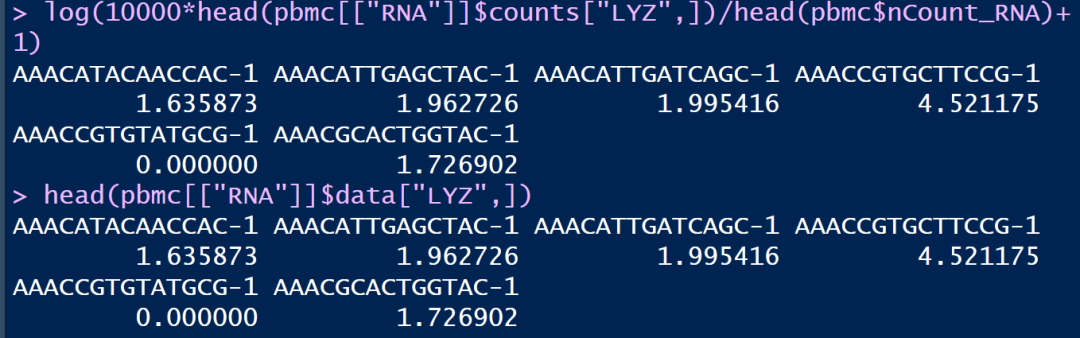
可以简单对比一下我们的计算以及使用NormalizeData()函数运行的结果
x=head(pbmc[["RNA"]]$data["LYZ",],100)
y=log(10000*head(pbmc[["RNA"]]$counts["LYZ",],100)/head(pbmc$nCount_RNA,100)+1)
plot(x,y)
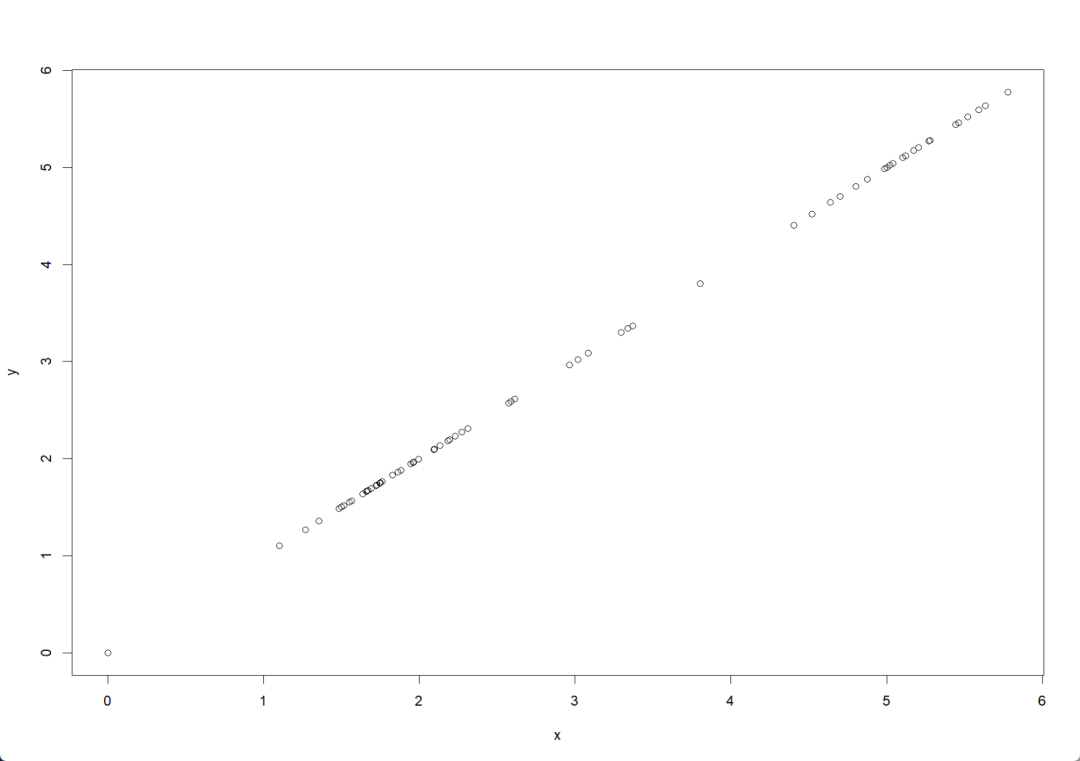
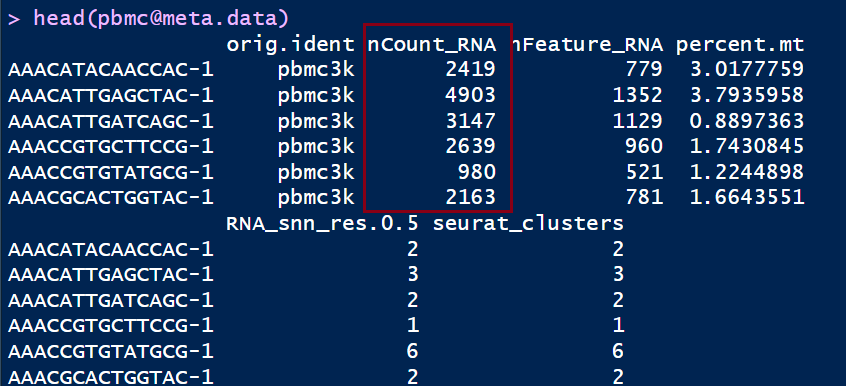
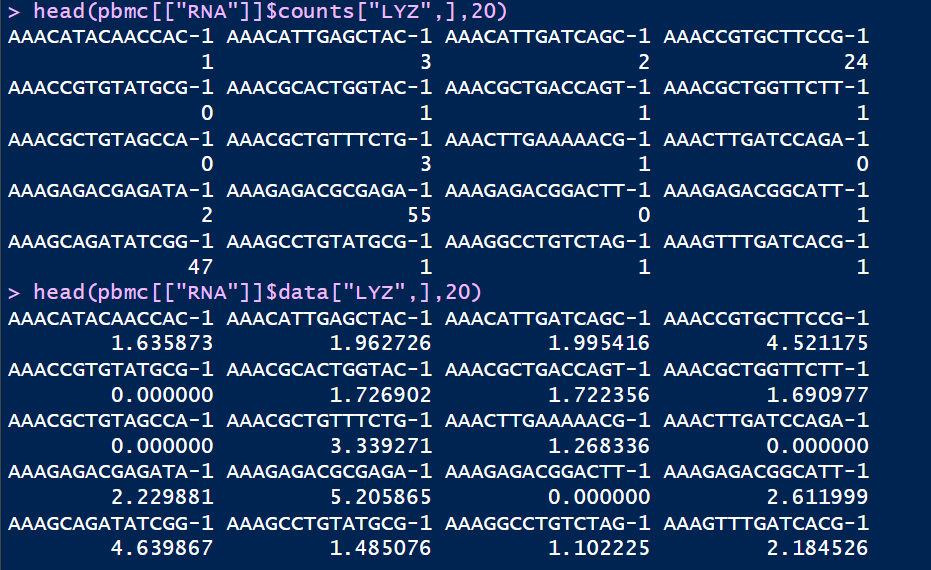
标准化后的数据存放在data数据栏,使用pbmc@assaysRNAdata可以查看,经过计算的data数据和原始的counts矩阵对比发现数值发生了变化
鉴定高变基因
高变基因:在某些细胞中高表达,而在其他细胞中低表达的基因
在分析时如果关注并使用这些基因进行后续的分析,那会节省分析的时间,并且有助于突出单细胞数据集中的生物信号
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
一般是默认计算并返回前2000个高变基因nfeatures = 2000,方法选用的是"vst"
“ vst ”:使用局部多项式回归 (loess) 拟合 log(方差)和 log(均值)关系的线,再使用观测到的均值和期望方差(由拟合线给出)对特征值进行标准化。然后,在裁剪到最大值后,根据标准化值计算特征方差。
#可视化一下TOP基因
top20 <- head(VariableFeatures(pbmc), 20)
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top20, repel = TRUE)
plot1 + plot2
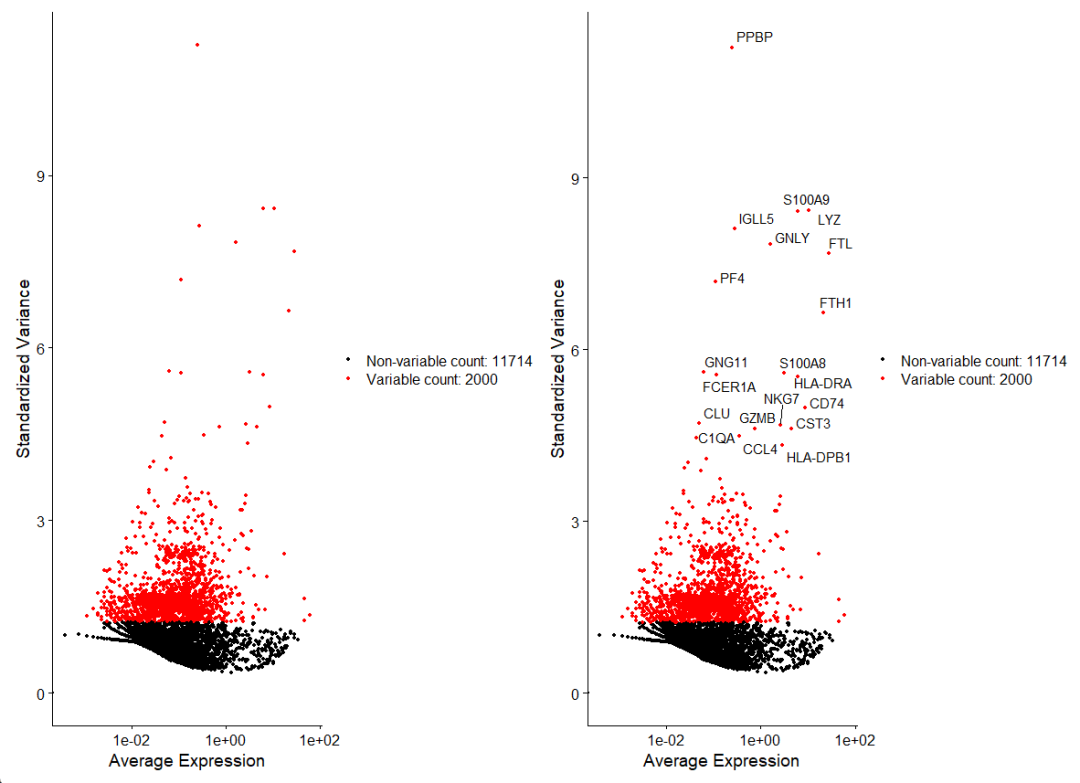
根据TOP20的高变基因我们也可以大概了解一下有哪些细胞类群,比如S100A9和S100A8就是monoctyes(单核细胞)的Marker基因
数据缩放
使用ScaleData()对数据进行线性变换,一般是对基因表达量处理
改变每个基因的表达,使细胞间的均值为0 缩放每个基因的表达,使细胞间的方差为1
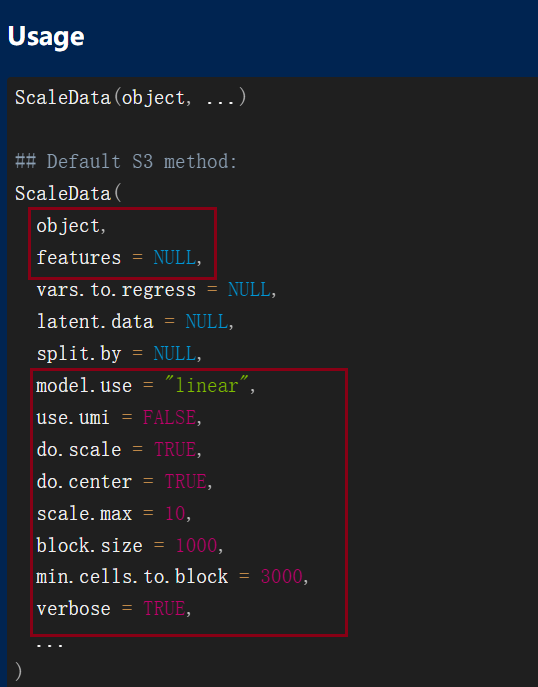
一般只需要指明我们的object以及进行缩放的features,如果不指明features的话,默认使用我们刚刚计算的高变基因
#对全部基因进行缩放
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
#对高变基因进行缩放
pbmc2 <- ScaleData(pbmc)
耗时的话,显而易见基于高变基因进行数据缩放时间会快一些,尤其是对于实际数据量大的情况
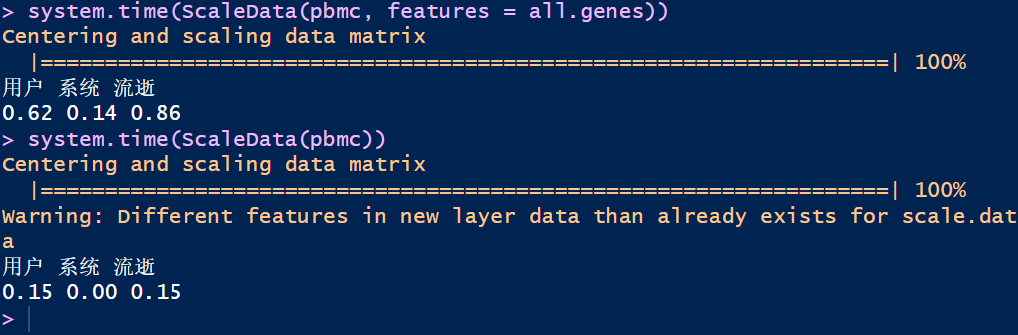
使用ScaleData()对数据进行缩放之后的值会保存在scale.data里面,使用pbmc[["RNA"]]$scale.data即可查看,通过ScaleData()会赋予每个基因在下游分析中同样的权重,不至于使高表达基因占据主导地位
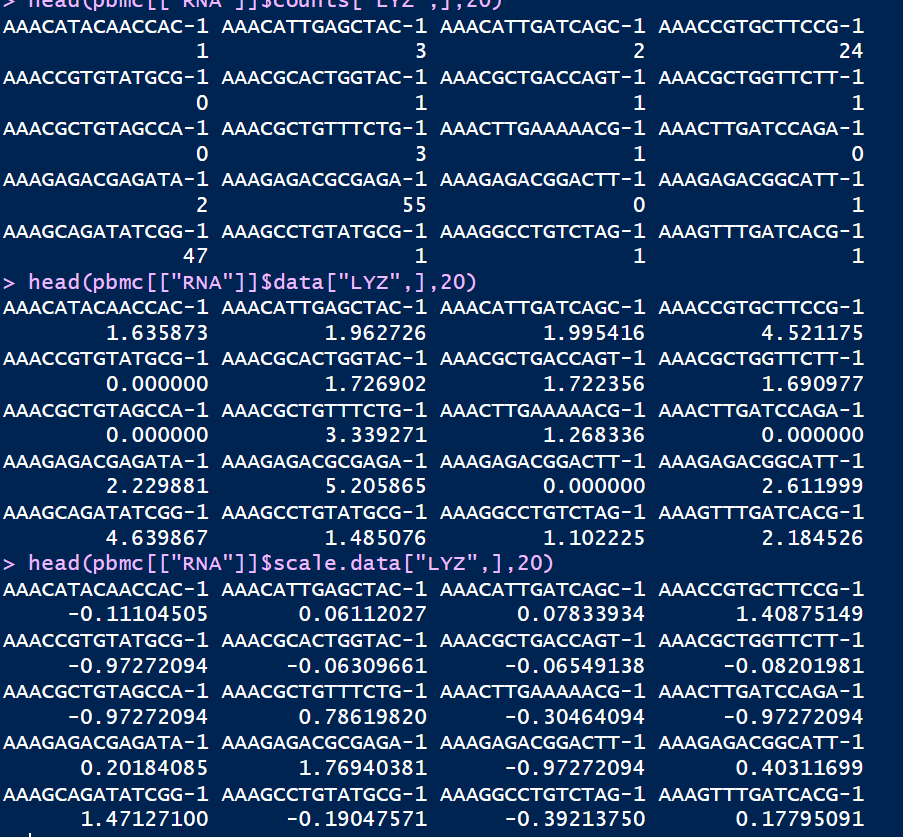
可以使用对scale.data进行简单的统计查看
rowSums(pbmc[["RNA"]]$scale.data)
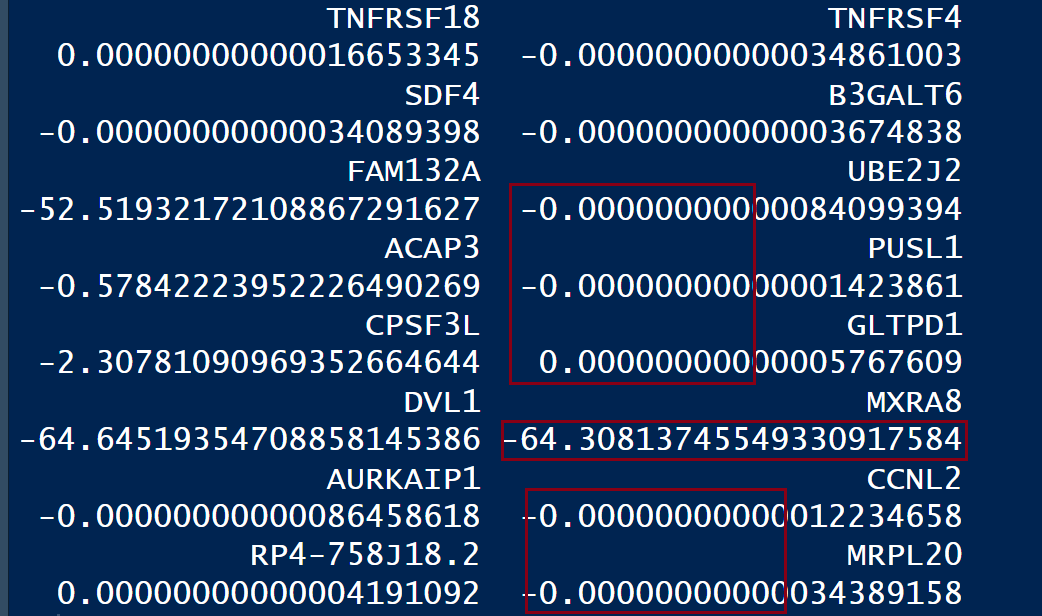
可以发现大部分基因都是无限接近于0的,这些基因在细胞中的counts表达量基本不为0
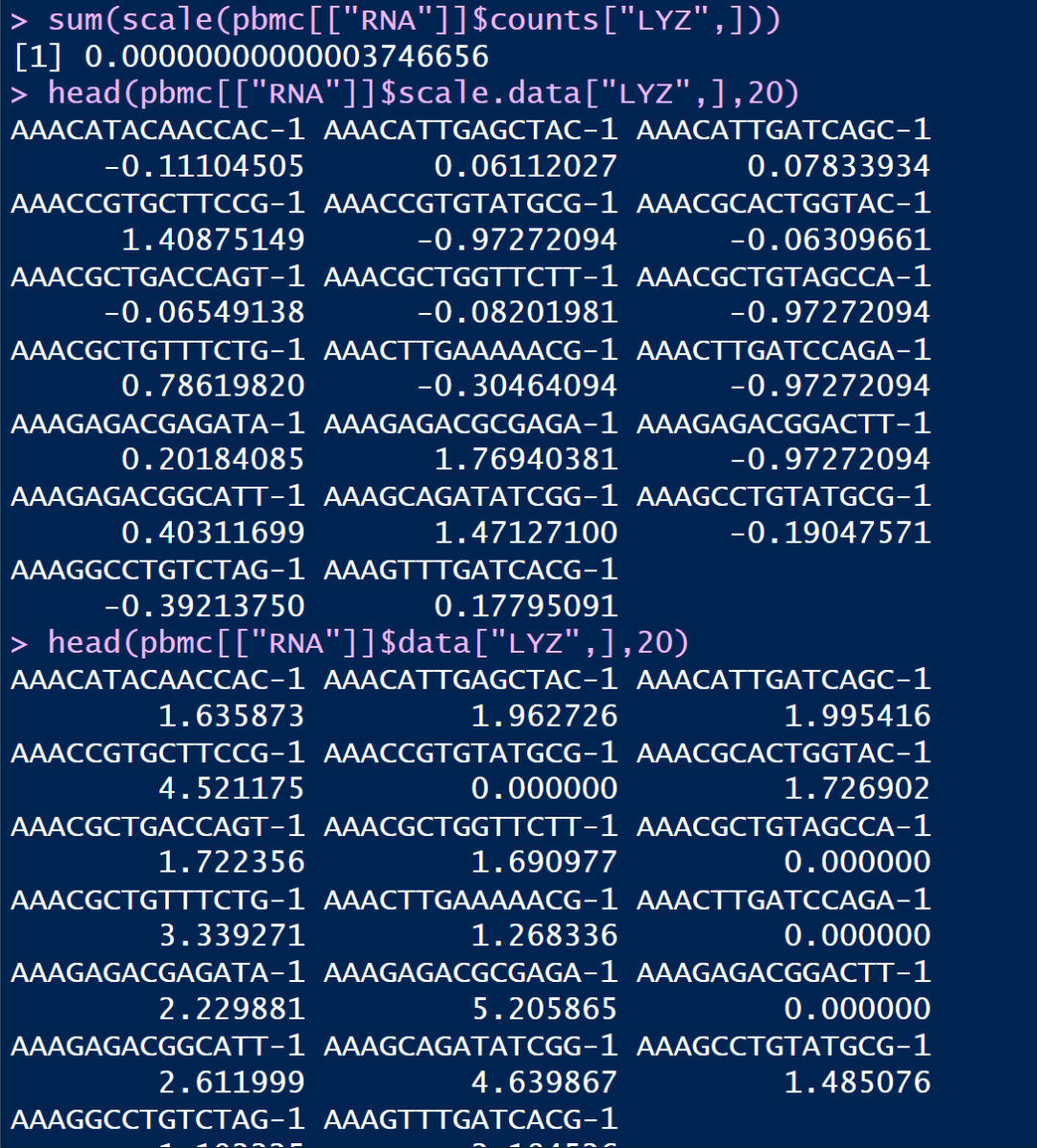
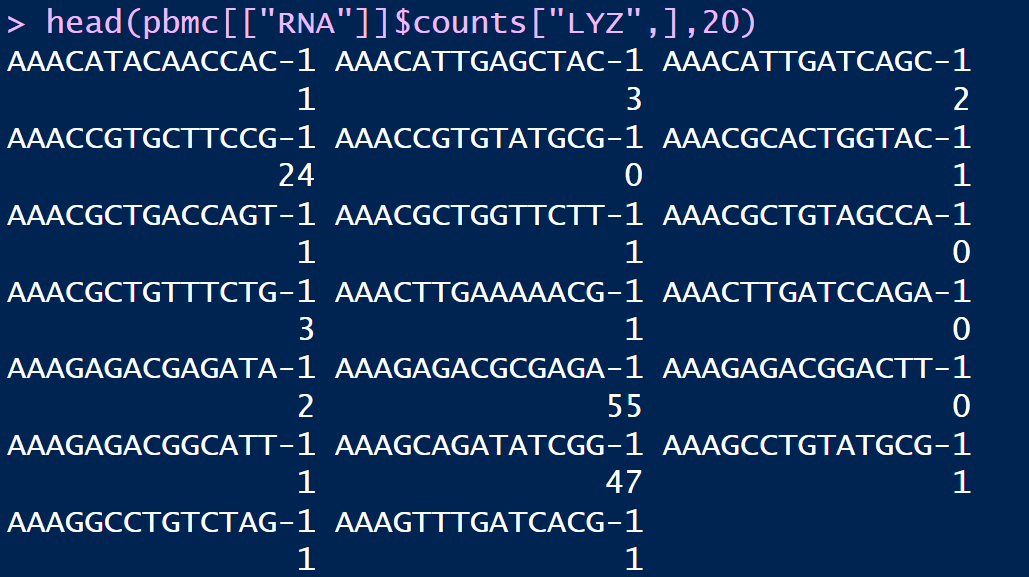
但有少部分基因是明显的负值,通过查看其表达量情况,可以发现该基因大部分的counts表达量都为0,这就导致在ScaleData()的时候没有很好的归一化