统计遗传学:第九章,GWAS+群体分析+亲缘关系分析

统计遗传学:第九章,GWAS+群体分析+亲缘关系分析

邓飞
发布于 2022-12-12 20:33:44
发布于 2022-12-12 20:33:44
本篇,使用数据和代码演示的形式,展示了GWAS分析、群体结构分析、亲缘关系分析三部分内容。我又重演了一遍,修正了一些bug。文中代码和数据我回头专门整理相关博文进行分享。
主要内容:
* PLINK软件进行GWAS分析
* LD筛选
* PCA分析
* PLINK软件计算IBS矩阵
* GCTA进行GWAS分析
* GCTA进行遗传力计算
大家好,我是飞哥,本章节是理论+实操,干货满满,这里我将书中的数据用代码进行了实现,你可以下载相关的数据,用我整理好的代码进行操作,666!
本书其它章节介绍
群体分析+亲缘关系分析
主要内容

本章节包括:
- 学习如何进行线性和logistic关联分析
- 了解如何进行加性、显性、,或对所选单核苷酸多态性(SNPs)进行隐性分析
- 能够使用PLINK进行全基因组关联研究
- 了解如何通过连锁不平衡(LD)修剪来识别独立SNPs
- 使用LD找到具有感兴趣SNP的高LD SNPs的代理基因型。
- 解如何在遗传数据中执行主成分分析
- 计算基因相关性使用PLINK和全基因组复杂性状分析(GCTA)的状态同一性(IBS)
- 使用GCTA估计不同表型的遗传力
简介
本章要点
前一章为读者提供了如何使用PLINK的一些基础知识,以及数据管理和质量控制的一些基础知识。本章的目的是向读者介绍关联分析、人口分层和遗传相关性的要点。在关联分析中检验SNP与特定性状之间的关系是许多分析的基础。然后,我们通常感兴趣的是检查SNP和特征之间的特定相关性是否来自独立或冗余的SNP。因此,我们说明了如何通过LD剪枝技术分离独立SNP。这是通过寻找与你感兴趣的特定SNP具有高LD的SNP来实现的。我们从第三章中了解到,不同祖先群体的个体在等位基因频率方面有所不同。因此,我们演示了如何计算遗传数据中的主成分。处理遗传数据时需要考虑的另一个重要因素是遗传相关性以及重复或相关个体可能带来的偏差。我们解释了如何使用PLINK和GCTA(全基因组复杂性状分析)中的状态同一性度量(IBS)来识别这些相关个体。我们向读者介绍了GCTA的一些基本命令,并提供了如何使用该程序计算遗传力的说明。
数据介绍
为了积极遵循本章,您需要确保将以下数据安装在我们将在本章中使用的适当目录中。请记住,本书中使用的数据见附录2,相关数据可从我们的配套网站下载,http://www.intro-statistical-genetics.com.除了。txt fle,都有链接的(.bed、.bim和.fam)文件:

注意,这里我已经将需要的示例数据下载了,目录如下:
第九章的数据和第八章的数据一致。
需要提前下载安装GCTA软件。
GWAS关联分析
常见的命令,在不同系统下的代码如下:
正如你从第一章所知道的,遗传分析的主要目标是估计基因型和表型之间的关联。作为下面的一个简单示例,我们估计rs9674439等位基因与体重指数(BM)的线性关联。统计模型估计C等位基因(bim文件中的第一个等位基因)对感兴趣表型的影响。到目前为止,这种模型是全基因组关联研究(GWASs)中最常见的。该SNP的C等位基因的每个拷贝具有相同的效果,或者换句话说。 这是一个相加模型。对定量表型具有相加效应的基本线性回归可以估计如下
命令及日志如下:
plink --bfile 1kg_EU_BMI --snps rs9674439 --assoc --linear --out BMIrs9674439
$ plink --bfile 1kg_EU_BMI --snps rs9674439 --assoc --linear --out BMIrs9674439
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to BMIrs9674439.log.
Options in effect:
--assoc
--bfile 1kg_EU_BMI
--linear
--out BMIrs9674439
--snps rs9674439
15236 MB RAM detected; reserving 7618 MB for main workspace.
851065 variants loaded from .bim file.
379 people (178 males, 201 females) loaded from .fam.
379 phenotype values loaded from .fam.
--snps: 1 variant remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 379 founders and 0 nonfounders present.
Calculating allele frequencies... done.
1 variant and 379 people pass filters and QC.
Phenotype data is quantitative.
Writing QT --assoc report to BMIrs9674439.qassoc ... done.
Writing linear model association results to BMIrs9674439.assoc.linear ... done.
结果:
$ head BMIrs9674439.assoc.linear
CHR SNP BP A1 TEST NMISS BETA STAT P
16 rs9674439 33836510 C ADD 379 -0.2974 -1.269 0.2052
在处理病例对照研究时,回归略有不同,应省略线性选项。计算的统计检验为卡方检验,估计系数为优势比。通过将PLINK命令中的-logistic替换为-1linear,还可以使用二元表型的逻辑回归选项。在下面的例子中,我们对二元特征(超重)进行了逻辑回归。体重指数大于或等于25的个体被归类为超重(病例),而体重指数小于25的个体被归类为不超重(对照)。在PLINK中,病例编码为2,对照组编码为1。在这里,我们使用逻辑回归来估计rs9674439对超重概率的影响。
上面需要对表型数据进行重新编码,BMI小于25的编码为1(对照),大于等于25的编码为2(case,病例),命名为:1kg_EU_Overweight为前缀的二进制文件。
cp 1kg_EU_BMI.bed 1kg_EU_Overweight.bed
cp 1kg_EU_BMI.fam 1kg_EU_Overweight.fam
cp 1kg_EU_BMI.bim 1kg_EU_Overweight.bim
上面的代码,只有379个个体,还是要通过原始数据进行更新:
plink --bfile 1kg_hm3_QC --pheno BMI_pheno.txt --make-bed --out 1kg_EU_Overweight
R代码,处理生成Overweight文件:
library(tidyverse)
library(data.table)
fam = fread("1kg_EU_Overweight.fam")
head(fam)
summary(fam)
fam = fam %>% mutate(V6 = case_when(
V6 >=25 ~ 2,
V6 < 25 ~ 1))
summary(fam)
fwrite(fam,"1kg_EU_Overweight.fam",sep = " ",col.names = F,na = -9,quote = F)
代码:
plink --bfile 1kg_EU_Overweight --snps rs9674439 --assoc --logistic --out BMIrs9674439
日志:
$ plink --bfile 1kg_EU_Overweight --snps rs9674439 --assoc --logistic --out BMIrs9674439
plink --bfile 1kg_EU_Overweight --snps rs9674439 --assoc --logistic --out BMIrs9674439
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to BMIrs9674439.log.
Options in effect:
--assoc
--bfile 1kg_EU_Overweight
--logistic
--out BMIrs9674439
--snps rs9674439
15236 MB RAM detected; reserving 7618 MB for main workspace.
846484 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
1092 phenotype values loaded from .fam.
--snps: 1 variant remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 1083 founders and 9 nonfounders present.
Calculating allele frequencies... done.
1 variant and 1092 people pass filters and QC.
Among remaining phenotypes, 557 are cases and 535 are controls.
Writing C/C --assoc report to BMIrs9674439.assoc ... done.
Writing logistic model association results to BMIrs9674439.assoc.logistic ...
done.
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to BMIrs9674439.log.
Options in effect:
--assoc
--bfile 1kg_EU_Overweight
--logistic
--out BMIrs9674439
--snps rs9674439
15236 MB RAM detected; reserving 7618 MB for main workspace.
846484 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
1092 phenotype values loaded from .fam.
--snps: 1 variant remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 1083 founders and 9 nonfounders present.
Calculating allele frequencies... done.
1 variant and 1092 people pass filters and QC.
Among remaining phenotypes, 557 are cases and 535 are controls.
Writing C/C --assoc report to BMIrs9674439.assoc ... done.
Writing logistic model association results to BMIrs9674439.assoc.logistic ...
done.
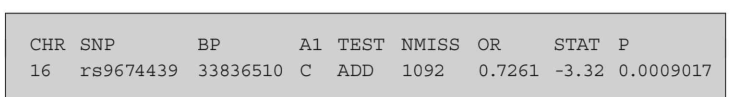
大多数读者都知道,逻辑回归的输出与线性回归略有不同。文件超重_rs9674439。logistic,如下所示有以下列:染色体数(CHR)、变体标识符(SNP)、碱基对位置(BP);效应等位基因(A1)、使用的统计检验类型(test)、缺失值数量(NMISS)、优势比(OR)、t统计量(STAT)和t统计量(p)的渐近p值。作为一个标准输出,PLINK报告了logistic回归的优势比估计,在本例中,该优势比是与C等位基因每个拷贝相关的超重概率与没有C等位基因拷贝的超重概率之间的比率。换句话说,它告诉我们,如果一个人至少有一个特定等位基因的拷贝(在加性模型中),那么他超重的可能性有多大。优势比总是大于零。当优势比大于l时,表明风险增加;当它小于0时,表示风险降低;当它等于1时,意味着没有关联。在下面的例子中,我们看到OR为0.7,表明C等位基因与超重概率降低有关。相关p值为0.0009,这意味着统计关联性很强。结果表明,rs0674439上有一个C等位基因可以防止超重。虽然统计显著性水平不同,但结果与之前的模型一致。这是因为我们在看同一个变量,但编码方式不同。体重指数是一个连续变量,而超重是两分的。因此,您对如何编码变量的选择非常重要,这取决于研究设计和您的研究问题。
结果可以书本中的结果一致。
$ cat BMIrs9674439.assoc.logistic
CHR SNP BP A1 TEST NMISS OR STAT P
16 rs9674439 33836510 C ADD 1092 0.7261 -3.32 0.0009017
加性模型是最常见的基因型-表型关联分析类型,尽管有时我们可能对不同的模型感兴趣,例如研究单个等位基因效应的显性模型或隐性模型。特别是,显性模型将杂合子和一个纯合子基因型视为一个单一类别。例如,如表9.1所示。让我们假设一个给定的SNP具有等位基因a和B。这三个可能的基因型组是AA。AB和BB。如果A是效应或“风险”等位基因,那么显性模型将研究至少有一个A拷贝的效应,即“AA+AB”与“BB”的效应相反,隐性模型估计具有两个a拷贝的效果,即“AA”与“AB+BB”的效果对于线性模型,可以在PLINK中使用选项(1近似显性或线性隐性)估计这些模型,如下例所示。

代码:
plink --bfile 1kg_EU_BMI --snps rs9674439 --assoc --linear dominant --out BMIrs9674439
结果:
$ cat BMIrs9674439.assoc.linear
CHR SNP BP A1 TEST NMISS BETA STAT P
16 rs9674439 33836510 C DOM 379 -0.4783 -1.462 0.1445
$ cat BMIrs9674439.qassoc
CHR SNP BP NMISS BETA SE R2 T P
16 rs9674439 33836510 379 -0.2974 0.2343 0.004254 -1.269 0.2052
PLINK中的关联分析可能包括协变量,如受访者性别、出生年份、人口分层控制或数据特定变量。在线性模型中,这可以通过添加--covar选项,然后添加一个选项卡分隔的文件来指定,该文件包括在分析中用作协变量的变量。在这种情况下,对于每个标记,输出文件将包括一行,指示模型中包括的每个协变量的回归估计。 如果我们对测试与基因型文件中包含的所有基因变体的关联感兴趣,而不是测试单个变体,那么可以通过省略--snp命令来实现。这是一种全基因组分析(即GWAS),第4章对此进行了广泛讨论,尽管这种分析通常包括基因型和插补数据。
代码:
plink --bfile 1kg_EU_BMI --assoc --linear --out BMIgwas
结果:
$ head BMIgwas.assoc.linear
CHR SNP BP A1 TEST NMISS BETA STAT P
1 rs1048488 760912 C ADD 379 0.6031 2.151 0.03208
1 rs3115850 761147 T ADD 379 0.6056 2.135 0.03343
1 rs2519031 793947 G ADD 379 -0.9188 -1.019 0.3087
1 rs4970383 838555 A ADD 379 -0.01473 -0.05882 0.9531
1 rs4475691 846808 T ADD 379 -0.3347 -1.221 0.223
1 rs1806509 853954 C ADD 379 -0.1015 -0.4786 0.6325
1 rs7537756 854250 G ADD 379 -0.1289 -0.4769 0.6337
1 rs28576697 870645 C ADD 379 0.1739 0.7539 0.4514
1 rs7523549 879317 T ADD 379 0.1316 0.2271 0.8204
PLINK中GWA的输出与单个回归的输出完全相同,并具有以下列:染色体数(CHR)、变异标识符(SNP)、碱基对位置(BP)、效应等位基因(A1)、使用的统计测试类型(测试)、缺失值数(NMISS)、回归系数(β)、t统计量(STAT)和t统计量(p)的渐近p值。 对于PLINK文件中包含的每个SNP,依次重复回归模型。如第4章所述,在解释GWAS结果时,我们需要考虑多重测试,以避免增加误报的数量。为了说明这一点,如果我们采用0.05的normalp值阈值,我们预计在零下。5.变异与偶然性显著相关。当我们仅使用几个感兴趣的变量估计回归时,这在许多情况下可能是可以接受的。当我们同时测试100万个变量(SNP)时,p值为0.05意味着50000个SNP为假阳性。为了避免这种错误,我们采用了更严格的p值阈值(5×10-8,即0.0000000.5)。 当使用GWAS结果时,您会注意到的另一个方面是,具有相似位置的SNP将具有相似的效果和p值。这是因为单核苷酸多态性在LD中(见第3章第3.6节)。我们将在第12章进一步介绍如何使用GWAS结果。
PLINK可以执行多种不同类型的关联分析。例如,可以进行家庭内分析(也称为家庭固定效应回归),在其中我们检查家庭成员之间不同基因型的影响。由于等位基因是通过减数分裂随机传播的,因此兄弟姐妹之间的基因型差异是一个真正的随机实验,因此家庭内分析对于建立不受人口分层和遗传养育影响的真正遗传效应至关重要(见第1章第1.2节)。这种分析可以在PLINK中使用命令gfam执行。更高级的关联分析也可以使用我们在本入门教材中没有介绍的PLINK进行,包括分层病例/对照分析、使用剂量数据的回归、套索回归和线性混合模型关联GWAS通常使用PLINK以外的软件进行,主要原因是,PLINK的当前版本并不最适合使用具有插补不确定性的数据。用于GWASs的其他软件包括SNPTEST、BOLT-LMM和BGENIE。目前正在开发的PLINK 2.0将有效地管理估算数据,并适用于大型GWASs。
连锁补平衡分析
如第3章(第3.6节)所述,连锁不平衡是许多因素的结果,如选择、遗传重组、突变率、遗传漂移、种群分层和遗传连锁。LD影响等位基因在人群中的分布方式,并在SNP之间创建相关结构。因此,我们通常对研究和检测SNP之间的相关性或识别独立的SNP感兴趣。基于几个原因,建立SNP之间的相关性是有用的。首先,在多项研究中可能无法测量相同的SNP。因此,即使在所有研究或子研究中没有测量到相同的SNP,我们也希望分离出遗传信号。第二个原因是为了减少后续分析的遗传变异数量。例如,如果我们想获得祖先信息,我们不需要基因型数据中包含的所有信息,因为其中大部分是冗余的。正如我们在下一章中所示,在计算多基因风险评分时,通常只提取独立的SNP。
单核苷酸多态性之间的LD通常用两种方法测量∶ r2和D。r测量值简单地计算为两个SNP之间等位基因相关系数的平方,因此它只能取0到1之间的值。它是两个标记之间共享信息的统计度量,通常用于确定一个SNP作为另一个SNP的代理的效果。统计D’是一种群体遗传学度量,也在0到1之间缩放,表示标记之间的重组概率。D等于0表示完全连锁平衡和频繁重组,而D'1表示两个标记之间没有重组,表示完全LD。PLINK还可用于检查两个标记之间的LD。选项——ld更详细地检查了一对变体之间的关系,第9224章显示了每个单倍型的观察和预期(基于MAFs)频率,以及基于单倍型的r2和D!
代码和结果:
plink --bfile hapmap-ceu --ld rs2883059 rs2777888 --out ld_example
日志:
$ plink --bfile hapmap-ceu --ld rs2883059 rs2777888 --out ld_example PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to ld_example.log.
Options in effect:
--bfile hapmap-ceu
--ld rs2883059 rs2777888
--out ld_example
15236 MB RAM detected; reserving 7618 MB for main workspace.
2239392 variants loaded from .bim file.
60 people (30 males, 30 females) loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 60 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.992022.
2239392 variants and 60 people pass filters and QC.
Note: No phenotypes present.
--ld rs2883059 rs2777888:
R-sq = 0.715909 D' = 1
Haplotype Frequency Expectation under LE
--------- --------- --------------------
CA -0 0.21
TA 0.45 0.24
CG 0.466667 0.256667
TG 0.083333 0.293333
In phase alleles are CG/TA
Warning: 59 het. haploid genotypes present (see ld_example.hh ); many commands
treat these as missing.
在这里,我们发现r2为0.715909,这表明rs2883059和rs2777888之间有相当高的相关性。D’等于1,这意味着这两个SNP处于完全LD中,或者换句话说,大约100%的时间是共同遗传的。不平衡值是两个或多个位点上等位基因非随机关联的度量。如果这两个基因座是独立的(例如,不是共同遗传的),那么无论哪个等位基因频率如何,r值和D值都将为0。其他人注意到,D’值受到上限效应的影响,或者换句话说,很容易达到1。但是你应该使用哪种方法呢?通常情况下,如果你的研究重点是一个多态性的可预测性,r2是首选的衡量标准。这就是为什么它经常用于关联设计的功率研究。 D是用于评估重组模式的度量,因为单倍型块通常被定义为D的基础,如上面的输出所示。 在某些情况下,我们可能有兴趣找到一个代理genotvpe,或者换句话说。 高LD的SNP与感兴趣的SNP。寻找代理基因型的最佳方法是检查参考面板,例如1000个基因组,记住不同的祖先群体可能在其LD结构上有很大差异。这可以通过使用在线数据库(如LDlink)来实现(https://ldlink.ncinihgov) [1].LDlink是一个包含多个基于web的应用程序的网站,旨在询问人口群体中的LD。表9.2报告了1000个基因组数据集中CEU群体3号染色体上SNP rs2777888的前10个代理基因型的LDlink结果。
LD Pruning
剪枝是一种统计过程,用于删除冗余SNP,或者换句话说,删除相关SNP对。通过迭代检查基因型数据中的所有SNP,LD剪枝只从每个LD块中选择一个代表性SNP。在每个步骤中,具有较高次要等位基因频率的SNP保留在数据集中。如果两个SNP的相关性()低于某个阈值,或者它们在碱基对中的距离大于指定值,则认为它们是独立的。例如,下面的命令告诉PLINK加载文件1kg hm3 qc,并保持SNP的MAF至少为1°??r2>0.2时,没有剩余对。默认情况下,相距超过1000千碱基的变体被认为是独立的。额外的参数,这里是50和5,会影响整个基因组的计算工作。!
代码及结果:
plink --bfile 1kg_hm3_qc --maf 0.01 --indep-pairwise 50 5 0.2 --out 1kg_hm3_qc_pruned
日志:
$ plink --bfile 1kg_hm3_qc --maf 0.01 --indep-pairwise 50 5 0.2 --out 1kg_hm3_qc_pruned PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to 1kg_hm3_qc_pruned.log.
Options in effect:
--bfile 1kg_hm3_qc
--indep-pairwise 50 5 0.2
--maf 0.01
--out 1kg_hm3_qc_pruned
15236 MB RAM detected; reserving 7618 MB for main workspace.
846484 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 1083 founders and 9 nonfounders present.
Calculating allele frequencies... done.
0 variants removed due to minor allele threshold(s)
(--maf/--max-maf/--mac/--max-mac).
846484 variants and 1092 people pass filters and QC.
Note: No phenotypes present.
Pruned 56392 variants from chromosome 1, leaving 13264.
Pruned 56216 variants from chromosome 2, leaving 12648.
Pruned 47468 variants from chromosome 3, leaving 11085.
Pruned 42986 variants from chromosome 4, leaving 10182.
Pruned 44973 variants from chromosome 5, leaving 10224.
Pruned 44379 variants from chromosome 6, leaving 10195.
Pruned 38436 variants from chromosome 7, leaving 9086.
Pruned 36869 variants from chromosome 8, leaving 8520.
Pruned 31703 variants from chromosome 9, leaving 7744.
Pruned 37089 variants from chromosome 10, leaving 8767.
Pruned 35953 variants from chromosome 11, leaving 8013.
Pruned 34449 variants from chromosome 12, leaving 8410.
Pruned 26600 variants from chromosome 13, leaving 6392.
Pruned 22218 variants from chromosome 14, leaving 5680.
Pruned 19268 variants from chromosome 15, leaving 5247.
Pruned 20156 variants from chromosome 16, leaving 5719.
Pruned 16819 variants from chromosome 17, leaving 5241.
Pruned 20241 variants from chromosome 18, leaving 5341.
Pruned 12170 variants from chromosome 19, leaving 4020.
Pruned 17071 variants from chromosome 20, leaving 4741.
Pruned 9577 variants from chromosome 21, leaving 2626.
Pruned 9323 variants from chromosome 22, leaving 2983.
Pruning complete. 680356 of 846484 variants removed.
Marker lists written to 1kg_hm3_qc_pruned.prune.in and
1kg_hm3_qc_pruned.prune.out .
上面的命令生成了一个独立SNP列表,可用于进一步分析。文件1kg\u hm3\u qcpruned。修剪in包含独立的SNP,而文件1kg\uhm3\u qc\u已修剪。修剪out列出了从修剪中排除的标记。为了获得“剪枝”数据集。我们可以在PLINK中使用--extract选项来去除冗余标记。
代码:换行用\,也可以去掉,变为一行代码,是一致的。多行更方便查看。
plink --bfile 1kg_hm3_qc \
--extract 1kg_hm3_qc_pruned.prune.in \
--make-bed \
--out 1kg_hm3_pruned
日志:
$ plink --bfile 1kg_hm3_qc \
--extract 1kg_hm3_qc_pruned.prune.in \
--make-bed \
--out 1kg_hm3_pruned
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to 1kg_hm3_pruned.log.
Options in effect:
--bfile 1kg_hm3_qc
--extract 1kg_hm3_qc_pruned.prune.in
--make-bed
--out 1kg_hm3_pruned
15236 MB RAM detected; reserving 7618 MB for main workspace.
846484 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
--extract: 166128 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 1083 founders and 9 nonfounders present.
Calculating allele frequencies... done.
166128 variants and 1092 people pass filters and QC.
Note: No phenotypes present.
--make-bed to 1kg_hm3_pruned.bed + 1kg_hm3_pruned.bim + 1kg_hm3_pruned.fam ...
done.
LD聚类是一种基于统计选择独立SNP的类似方法。例如,在计算多基因评分时,聚类的常见选择是使用从给定表型的GWA计算的检验统计量。通过这种方式,我们为每个LD区块的该遗传位点选择p值最小的SNP。我们将在第10章更详细地讨论LD聚集,该章演示了如何计算多基因分数。
群体结构分析
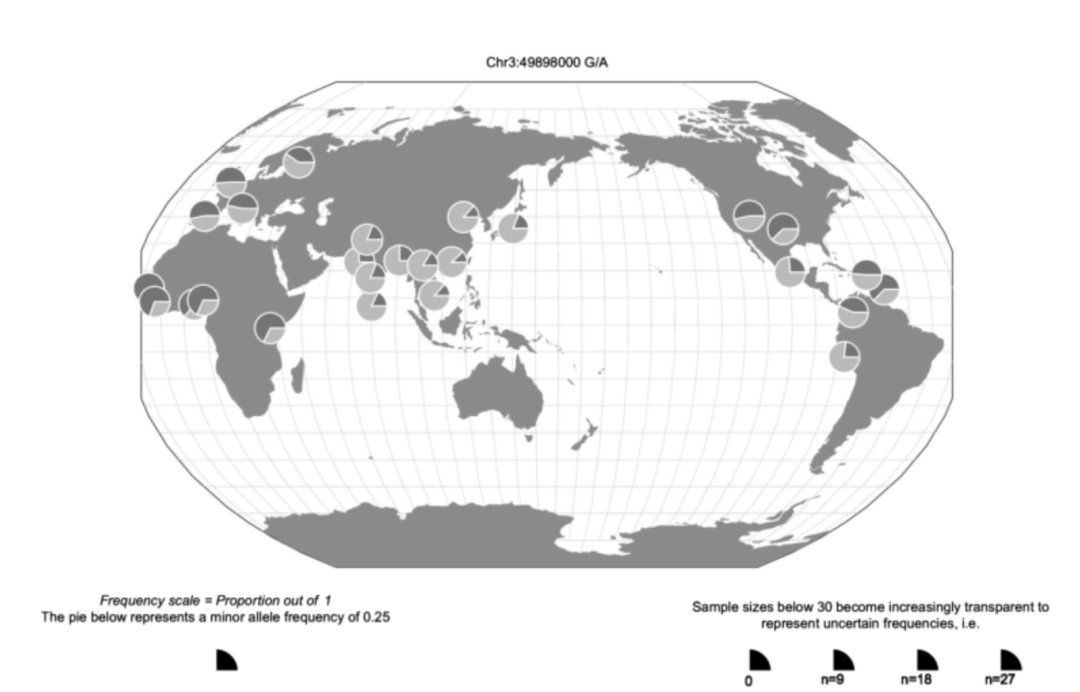
在第3章中讨论的,由于人类从非洲散居(第3.2节),来自不同祖先的个体在等位基因频率上存在显著差异。第3.3节详细阐述了人口结构和分层。在这里,我们注意到在一个群体中常见的单核苷酸多态性在另一个群体中可能很罕见,甚至根本没有变异。HapMap和1000个基因组等大型项目已经表明了遗传变异在不同群体中的变化。例如,图9.1显示了单核苷酸多态性rs2777888的等位基因频率,这是1000个基因组参考面板中与人类生殖行为密切相关的遗传标记。通过使用遗传变异地理浏览器[2],一个由芝加哥大学约翰·诺文布雷实验室提供的网络应用程序,我们可以绘制该SNP在不同人群中的变化。非洲人群中最常见的等位基因是G,而替代A等位基因在东南亚更为常见。 如第3章所述,人口分层对遗传关联有很大影响,在分析过程中必须仔细考虑。主成分分析(PCA)是识别和验证个体间祖先差异最广泛使用的方法。主成分分析是一种用于数据缩减的统计技术,用于将多维数据汇总到更少的变量中。通过从多元数据集计算主成分,我们降低了数据的复杂性,以说明原始数据集的结构。主成分分析用于遗传学解释个体样本中等位基因频率的差异。因此,主成分是解释原始数据中部分可变性的“新变量”。
主成分的重要性质是其内在排序。第一个组件总是具有最大解释价值的组件。然后是第二个,以此类推。在分析中通常使用遗传数据集的前10或20个主成分。如第3章第3.3.4节所述。遗传学中的主成分分析几乎完美地反映了不同群体的地理差异。主成分用于了解个体的祖先。此外,它们还用于OC,从样本中去除祖先异质的个体。最后,主成分分析是GWAS中用于纠正人口分层的标准方法之一。正如我们在第4章前面详细讨论的那样,GWAS目前通常专注于单个祖先群体,然后在另一个群体中复制(不幸的是,这意味着欧洲祖先的比例过高)[3]。然而,这不足以解释同一祖先群体内的进一步人口分层。例如,北欧个体的等位基因频率不同于南欧个体。可以使用几个软件包从遗传数据中估计主成分。其他程序可用于从遗传数据计算PCs,包括EIGENSTRAT。 在这里,我们报告了可以在PLINK中使用的命令,以使用选项pca 10估计前10个主成分。
代码:
./plink --bfile 1kg_hm3_pruned --pca 10 --out 1kg_pca
日志:
$ plink --bfile 1kg_hm3_pruned --pca 10 --out 1kg_pca
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to 1kg_pca.log.
Options in effect:
--bfile 1kg_hm3_pruned
--out 1kg_pca
--pca 10
15236 MB RAM detected; reserving 7618 MB for main workspace.
166128 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
Using up to 8 threads (change this with --threads).
Before main variant filters, 1083 founders and 9 nonfounders present.
Calculating allele frequencies... done.
166128 variants and 1092 people pass filters and QC.
Note: No phenotypes present.
Relationship matrix calculation complete.
--pca: Results saved to 1kg_pca.eigenval and 1kg_pca.eigenvec .
PLINK中的-pca命令生成两个输出文件。在本例中:1kg_pca。 eigenval和1kg PCA。特征向量。扩展名为的文件。特征向量是主成分列表,可由其他统计软件用于进一步分析。1kg pca的摘录。特征值文件为:
结果文件:
特征值:
$ head 1kg_pca.eigenval
54.1464
40.0338
6.96377
3.375
2.75908
2.16491
1.99634
1.99175
1.90434
1.87444
特征向量:
$ head 1kg_pca.eigenvec
0 HG00096 0.0149253 -0.0329941 0.0157409 0.00171199 0.00178966 -0.00704659 -0.00461685 -0.00735375 -0.00169564 0.0100253
0 HG00097 0.0146554 -0.0330726 0.0168457 -0.00070785 -0.000456348 -0.00860046 -0.00610165 -0.00293391 0.00189605 0.00350145
在计算主成分后,我们可以使用R检验个体在这些维度上的差异。使用RStudio,我们可以打开PLINK输出文件并直观地检查数据。图9.2是根据PLINK计算的两个第一主成分的散点图。我们可以立即认识到,数据不是随机分布的,而是从数据中出现了几个组(图9.2,上部面板)。 然后,我们可以使用1000个基因组样本中的信息来识别数据中的不同组。文件1公斤样本。txt包括1000个基因组样本中所有个体的起源种群。使用RStudio,我们可以导入该数据集并与PLINK计算的主成分合并,下面的代码生成一个更新的图,我们可以轻松区分数据中的每个群体。具有欧洲血统的个体聚集在图的右下角,东亚人聚集在图的右上角。非洲血统的个体在X轴上更为分散(PC 1),而美国人口在第二主成分上更为分散。
R语言作图:
Panel A
1ibrary(ggplot2)
columns=c("fid","Sample.name","pca1","pca2","pca3",
"pca4","pca5","pca6","pca7","pca8","pca9","pcal0")
pca<-read.table(file="1kg_ pca.eigenvec", sep = "",
header=F,col.names=columns)[,c(2:12)]
ggplot(pca, aes(x=pcal,y=pca2))+ geom_point()+
theme bw()+ xlab("PCI")+ ylab("PC2")
#Panel B
geo <- read.table(file="1kg_samples.txt",
sep = "\t",header=T)[,c(1,4,5,6,7)]
ata <- merge(geo,pca,by= "Sample.name")
ggplot(data, aes(x=pcal,y=pca2,col=Superpopulation.
name))+
geom _ point()+ theme bw()+
xlab("PC1")+ ylab("PC2")+
labs(col= "")
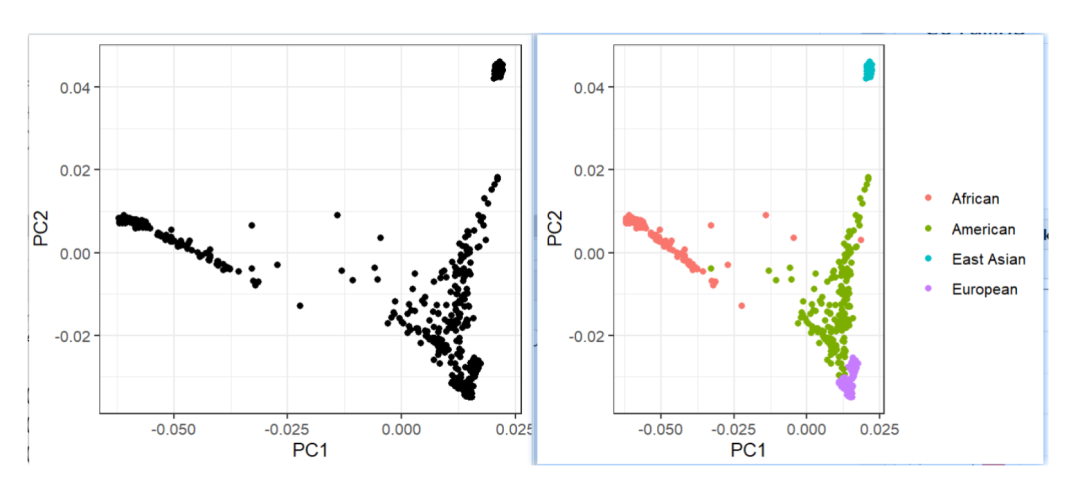
图9.3显示了一个图矩阵,表示1000个基因组样本之间的差异。有趣的是,主成分分析既可以区分祖先群体中的宏观差异,也可以区分更同质群体中的较小细节。将我们的分析局限于一个祖先群体,例如具有欧洲血统的个人,并不能保护我们免受由于人口分层而在分析中包含偏见的风险。例如,我们重复相同的情节,但这一次只针对具有欧洲血统的个人(图9.4)。虽然不同组之间的差异不如前一个图中的显著,但通过比较不同的主成分,可以立即看出如何区分不同的组。(本教科书仅显示所有数字的灰度版本,彩色图显示在本书的在线同伴网站上)主成分分析已被证明是一种非常有效的方法,用于识别不同祖先的个体,并在分析中控制人口分层。最近,GWASs中还引入了其他方法来解释人口分层的差异。最有效的方法是使用家庭固定效应模型,即基于对DZ(双卵非同卵)双胞胎或正常兄弟姐妹的分析。兄弟姐妹之间的遗传差异是由于减数分裂引起的随机性,根据定义,生物兄弟姐妹之间没有种群分层。对兄弟姐妹进行关联比较是估计遗传变异“实际影响”的有效方法。然而,这种方法是有代价的。由于兄弟姐妹平均分享50%的遗传变异,许多SNP在兄弟姐妹之间具有完全相同的等位基因。这意味着,如果我们想要使用同胞差异来检测与不相关个体群体相同的细节水平,则需要更大的样本。遗传学家收集了许多基于家庭的研究,其中兄弟姐妹和双胞胎都有基因型。然而,这些研究的样本量远小于较大的数据倡议,如英国生物银行[4]。家庭固定效应模型通常被用作稳健性检查,以验证在较大人口中获得的主要结果。然而,一个问题是,小家庭可能被系统地排除在这些类型的分析之外。关联研究中越来越多地采用的另一种方法是使用线性混合模型而不是主成分分析来控制人口分层。线性混合模型是考虑数据中遗传相关程度的回归模型。这类模型首先估计遗传相关性矩阵(见下一节),并将此信息包含在回归模型中。软件BOLT-LMM[5]使用这种方法来执行GWASs。
亲缘关系分析
如前一章所述,重复和相关个体在关联分析中会显著引入偏差。为了识别基因型数据中的相关个体,可以根据每对个体共同拥有的共享等位基因的平均比例来计算称为状态认同(BS)的度量。IBS通常根据一组独立的基因型SNP计算。我们可以使用PLINK根据个体之间基因型SNP的差异计算IBS,使用选项-距离。
数据中,没有EUR数据,用下面命令生成:
grep EUR 1kg_samples.txt |awk '{print 0,$1}' >1kg_samples_EUR.txt
代码:
plink --bfile 1kg_hm3_pruned --keep 1kg_samples_EUR.txt --distance --out ibs_matrix
日志:
$ plink --bfile 1kg_hm3_pruned --keep 1kg_samples_EUR.txt --distance --out ibs_matrix
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to ibs_matrix.log.
Options in effect:
--bfile 1kg_hm3_pruned
--distance
--keep 1kg_samples_EUR.txt
--out ibs_matrix
15236 MB RAM detected; reserving 7618 MB for main workspace.
166128 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
--keep: 379 people remaining.
Using up to 8 threads (change this with --threads).
Before main variant filters, 379 founders and 0 nonfounders present.
Calculating allele frequencies... done.
166128 variants and 379 people pass filters and QC.
Note: No phenotypes present.
Distance matrix calculation complete.
IDs written to ibs_matrix.dist.id .
Distances (allele counts) written to ibs_matrix.dist .
该命令生成一个以制表符分隔的文本文件,其中包含个体之间的汉明距离。文件组织如下。第一个元素是基因组1和基因组2之间的距离。基因组与自身之间的距离被忽略,因为它当然是0。默认命令生成下三角文件。事实上,IBS距离是对称的,因为基因组1和2之间的距离相当于基因组2和基因组1之间的距离。然而,如果我们使用命令——距离平方,就有可能生成一个正方形文件。1kg\uSamples\uEUR中前10个基因组的距离。此处显示txt文件。
结果文件:
$ head ibs_matrix.dist
74407
74208 74953
74755 74396 73099
74429 74572 73941 73346
74049 74752 74473 74218 74544
74792 74829 74704 75257 74797 74915
75499 74890 74743 74858 74740 74780 75025
74915 74522 74291 75122 74148 74892 73305 73548
74601 74286 74731 74774 74502 74734 75247 75260 74556
73911 74190 73761 74260 74132 74018 74595 74408 74186 74116
使用命令make rel给出了距离的替代度量,该命令使用GCTA软件使用的相同度量给出了遗传相关性矩阵。
代码:
plink --bfile 1kg_hm3_pruned --keep 1kg_samples_EUR.txt --make-rel --out rel_matrix
日志:
$ plink --bfile 1kg_hm3_pruned --keep 1kg_samples_EUR.txt --make-rel --out rel_matrix
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to rel_matrix.log.
Options in effect:
--bfile 1kg_hm3_pruned
--keep 1kg_samples_EUR.txt
--make-rel
--out rel_matrix
15236 MB RAM detected; reserving 7618 MB for main workspace.
166128 variants loaded from .bim file.
1092 people (525 males, 567 females) loaded from .fam.
--keep: 379 people remaining.
Using up to 8 threads (change this with --threads).
Before main variant filters, 379 founders and 0 nonfounders present.
Calculating allele frequencies... done.
166128 variants and 379 people pass filters and QC.
Note: No phenotypes present.
Relationship matrix calculation complete.
Relationship matrix written to rel_matrix.rel , and IDs written to
rel_matrix.rel.id .
这些结果可用于发现隐相关。MZ双胞胎或复制对的预期值为1;一级亲属(如全同胞或亲子)为0.5;二级亲属(如祖父母、孙子女)为0.25;三级亲属(如表亲)为0.125。请注意,这些是预期值,实证结果可能不同。例如,研究表明,尽管亲生兄弟姐妹之间的平均遗传距离为0.5,但差异的典型范围为0.4到0.6[6]。 通过观察下面的结果,我们可以注意到,例如,个体和自身的遗传相关性不同于1。这是基于个体内单倍型关系的近交效率估计[7]。然而,值得注意的是,一些个体对具有更高的遗传相关值。基因组4和基因组3在不相关个体中的遗传相关性高于预期。基于这个值,我们可以得出结论,这些人是二级亲属。同时,我们也可以看到一些单元格有负值。这是因为使用不相关个体之间的平均距离对遗传相关性值进行归一化。因此,仅在来自同一祖先群体的个体之间计算遗传相关矩阵非常重要。
查看结果:
$ head rel_matrix.rel
0.975623
0.00309471 0.980394
0.00433624 -0.00307206 0.977987
0.00320043 0.00425093 0.0261669 0.984606
0.00260946 0.00411425 0.0125727 0.0228138 0.982296
0.00847317 -0.00099271 -0.00231934 0.0101367 0.00190884 0.966281
0.00107227 -0.000977606 0.00169476 -0.00533466 0.0012784 0.00239035 0.987757
-0.00668254 0.00174476 0.00297298 0.00209255 0.00394602 0.00300734 0.00412087 0.996598
-0.00648276 0.00111926 0.00115654 -0.00728533 0.00529989 -0.00601775 0.0235142 0.0200166 0.963359
0.00560678 0.00777351 -0.000643365 0.00147696 0.00723983 0.000708483 -0.00598989 -0.00267611 0.000574517 0.986805
使用GCTA计算遗传力
另一种计算相关矩阵并进行额外分析的方法是使用软件GCTA(全基因组复杂性状分析),该软件由杨和澳大利亚昆士兰大学的同事开发。关于如何安装GCTA的说明包含在附录1中。GCTA是最初设计用于使用全基因组数据估计遗传力的软件,但后来扩展到执行许多其他分析。在本节中,我们简要介绍了GCTA在估计个体相关性和基于SNP的遗传力方面的一些基本应用。 GCTA是一种基于命令的软件(如PLINK),可以轻松处理PLINK二进制文件。计算基于SNP的遗传力的第一步是使用make GRM命令计算基因组关系矩阵(GRM)。下面我们提供了一个关于如何从二进制PLINK文件1kg\u EU\u qc估计GRM的示例。bim,1kg _uEU_uQC。床和1kg _EU _qC。fam。
代码:
gcta --bfile 1kg_EU_BMI --autosome --maf 0.01 --make-grm --out 1kg_gcta
日志:
$ gcta --bfile 1kg_EU_BMI --autosome --maf 0.01 --make-grm --out 1kg_gcta
*******************************************************************
* Genome-wide Complex Trait Analysis (GCTA)
* version 1.94.0 beta Windows
* (C) 2010-present, Jian Yang, The University of Queensland
* Please report bugs to Jian Yang <jian.yang.qt@gmail.com>
*******************************************************************
Analysis started at 15:48:27 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022.
Hostname: LAPTOP-0R4FUGN8
Options:
--bfile 1kg_EU_BMI
--autosome
--maf 0.01
--make-grm
--out 1kg_gcta
Note: GRM is computed using the SNPs on the autosomes.
Reading PLINK FAM file from [1kg_EU_BMI.fam]...
379 individuals to be included from FAM file.
379 individuals to be included. 178 males, 201 females, 0 unknown.
Reading PLINK BIM file from [1kg_EU_BMI.bim]...
851065 SNPs to be included from BIM file(s).
Threshold to filter variants: MAF > 0.010000.
Computing the genetic relationship matrix (GRM) v2 ...
Subset 1/1, no. subject 1-379
379 samples, 851065 markers, 72010 GRM elements
IDs for the GRM file have been saved in the file [1kg_gcta.grm.id]
Computing GRM...
100% finished in 12.1 sec
851065 SNPs have been processed.
Used 833298 valid SNPs.
The GRM computation is completed.
Saving GRM...
GRM has been saved in the file [1kg_gcta.grm.bin]
Number of SNPs in each pair of individuals has been saved in the file [1kg_gcta.grm.N.bin]
Analysis finished at 15:48:51 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022
Overall computational time: 24.55 sec.
老师1
参数介绍:

结果介绍:
$ ls 1kg_gcta.grm.*
1kg_gcta.grm.N.bin 1kg_gcta.grm.bin 1kg_gcta.grm.id
image-20220715122240861
第二步是从样本中删除相关个体。这可以通过从GRM中删除关系比指定水平更密切的个体对来实现。在下面的示例中,我们删除了关联度大于截止值0.025的个体,但请注意,保留了其中一个个体。这将创建一个新矩阵,称为1kgrm025。在这个例子之后,我们从分析中删除了59个个体,样本中只剩下320个。
命令:
gcta --grm 1kg_gcta --grm-cutoff 0.025 --make-grm --out 1kg_rm025
日志:
$ gcta --grm 1kg_gcta --grm-cutoff 0.025 --make-grm --out 1kg_rm025
*******************************************************************
* Genome-wide Complex Trait Analysis (GCTA)
* version 1.94.0 beta Windows
* (C) 2010-present, Jian Yang, The University of Queensland
* Please report bugs to Jian Yang <jian.yang.qt@gmail.com>
*******************************************************************
Analysis started at 15:57:08 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022.
Hostname: LAPTOP-0R4FUGN8
Options:
--grm 1kg_gcta
--grm-cutoff 0.025
--make-grm
--out 1kg_rm025
Pruning the GRM with a cutoff of 0.025000...
Total number of parts to be processed: 1
Processing part 1
After pruning the GRM, there are 320 individuals (59 individuals removed).
Pruned unrelated IDs have been saved to 1kg_rm025.grm.id
Pruning GRM values, total parts 1
Processing part 1
GRM values have been saved to [1kg_rm025.grm.bin]
Pruning number of SNPs to calculate GRM, total parts 1
Processing part 1
Number of SNPs has been saved to [1kg_rm025.grm.N.bin]
Analysis finished at 15:57:08 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022
Overall computational time: 0.01 sec.
第三步涉及估计SNP遗传力(h'e;与家族和GWAS遗传力相反;见第1章)。为了进行分析,我们需要提供表型。我们使用模拟的BMI变量(也在前一章中使用),并应用选项--pheno来提供包含表型的外部文件。
gcta --grm 1kg_rm025 --pheno BMI_pheno.txt --reml --out 1kg_BMI_h2
分析的总结结果已保存在一个名为1kg BMI h2的新文件中。hsq。
结果:
$ gcta --grm 1kg_rm025 --pheno BMI_pheno.txt --reml --out 1kg_BMI_h2
*******************************************************************
* Genome-wide Complex Trait Analysis (GCTA)
* version 1.94.0 beta Windows
* (C) 2010-present, Jian Yang, The University of Queensland
* Please report bugs to Jian Yang <jian.yang.qt@gmail.com>
*******************************************************************
Analysis started at 15:58:07 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022.
Hostname: LAPTOP-0R4FUGN8
Accepted options:
--grm 1kg_rm025
--pheno BMI_pheno.txt
--reml
--out 1kg_BMI_h2
Note: This is a multi-thread program. You could specify the number of threads by the --thread-num option to speed up the computation if there are multiple processors in your machine.
Reading IDs of the GRM from [1kg_rm025.grm.id].
320 IDs are read from [1kg_rm025.grm.id].
Reading the GRM from [1kg_rm025.grm.bin].
GRM for 320 individuals are included from [1kg_rm025.grm.bin].
Reading phenotypes from [BMI_pheno.txt].
Non-missing phenotypes of 1093 individuals are included from [BMI_pheno.txt].
320 individuals are in common in these files.
Performing REML analysis ... (Note: may take hours depending on sample size).
320 observations, 1 fixed effect(s), and 2 variance component(s)(including residual variance).
Calculating prior values of variance components by EM-REML ...
Updated prior values: 4.31259 4.31377
logL: -506.011
Running AI-REML algorithm ...
Iter. logL V(G) V(e)
1 -506.01 4.09746 4.52825
2 -506.01 3.57477 5.04950
3 -506.01 3.42696 5.19711
4 -506.01 3.38253 5.24145
5 -506.01 3.36894 5.25501
6 -506.01 3.36476 5.25918
7 -506.01 3.36347 5.26046
8 -506.01 3.36307 5.26086
Log-likelihood ratio converged.
Calculating the logLikelihood for the reduced model ...
(variance component 1 is dropped from the model)
Calculating prior values of variance components by EM-REML ...
Updated prior values: 8.62572
logL: -506.06662
Running AI-REML algorithm ...
Iter. logL V(e)
1 -506.07 8.62572
Log-likelihood ratio converged.
Summary result of REML analysis:
Source Variance SE
V(G) 3.363073 8.073311
V(e) 5.260860 8.059914
Vp 8.623934 0.683136
V(G)/Vp 0.389970 0.934748
Sampling variance/covariance of the estimates of variance components:
6.517835e+01 -6.483694e+01
-6.483694e+01 6.496221e+01
Summary result of REML analysis has been saved in the file [1kg_BMI_h2.hsq].
Analysis finished at 15:58:07 ▒й▒▒▒ʱ▒▒ on Mon Jul 18 2022
Overall computational time: 0.43 sec.
结果:
$ cat 1kg_BMI_h2.hsq
Source Variance SE
V(G) 3.363073 8.073311
V(e) 5.260860 8.059914
Vp 8.623934 0.683136
V(G)/Vp 0.389970 0.934748
logL -506.006
logL0 -506.067
LRT 0.122
df 1
Pval 3.6369e-01
n 320
GCTA将体重指数的方差分解为两部分:V(G),这是由加性遗传学解释的方差,和V(e),这是由环境引起的部分(非遗传部分)。遗传力(h'swp)的估计是V(G)占表型总方差(Vp)的比例。结果是0.39。因此,我们估计该表型(BMI)的变异几乎有40%归因于遗传学。 然而,遗传力的估计有一定程度的不确定性。h2w估计的精度由估计的标准误差(SE)表示。在这种情况下,SE为0.93,非常高,是点估计值的两倍以上。这意味着估计是不精确的。GCTA执行一项称为对数似然比测试的统计测试,该测试表明hsyp估计值与零,或者换句话说,与零假设不同。在这种情况下,假设值为0.36,我们无法拒绝零假设。因此,我们不能从这个例子得出结论,体重指数的遗传力不同于零,这意味着没有证据表明体重指数的遗传成分。事实上,体重指数的较大组成部分归因于遗传学,(大约20到30?但使用GCTA检测体重指数需要较大的样本量。 该示例仅基于我们320个个体的简单模拟测试样本,该样本太小,无法使用GCTA进行任何有意义的分析。在本章中,我们只能展示GCTA的一个非常基本的应用。使用该软件可以估计更复杂的模型,包括双变量模型或包含多个矩阵的模型,其中遗传方差分解为多个部分[7,8]。
结论
与第8章一起,本章为您提供了有关如何使用基因组数据的基本介绍。这些章节为我们将在本书最后一部分进行的更深入的遗传数据分析奠定了基础。在进行更详细的分析时,你会发现了解如何识别独立SNP和计算主成分、遗传相关性和遗传力很重要。下一章将说明如何创建多基因分数。然后在本书第二部分的后一个更高级的章节中应用。
练习

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-10-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号