如何使用plink进行二分类性状的GWAS分析并计算PRS得分

如何使用plink进行二分类性状的GWAS分析并计算PRS得分

邓飞
发布于 2022-12-13 17:35:52
发布于 2022-12-13 17:35:52
大家好,我是邓飞,PRS模型最近学了了不少内容。
这篇博客,用之前GWAS教程中的示例数据(快来领取 | 飞哥的GWAS分析教程),把数据分为Base数据和Target数据,通过plink运行二分类的logistic模型进行GWAS分析,然后通过PRSice-2软件,进行PRS分析。最终,选出最优SNP组合,并计算Target的PRS得分,主要结果如下:
最适合的SNP个数是133个,R2位0.232258,P值为0.014
$ head PRSice.summary
Phenotype Set Threshold PRS.R2 Full.R2 Null.R2 Prevalence Coefficient Standard.Error P Num_SNP
- Base 0.00115005 0.232258 0.232258 0 - 17.1644 6.99987 0.0142024 133
对应的43个个体的PRS结果:
$ head PRSice.best
FID IID In_Regression PRS
1328 NA06984 Yes 0.0386390029
13291 NA06986 Yes -0.136642922
1418 NA12272 Yes -0.140312974
1421 NA12287 Yes -0.154526751
1330 NA12340 Yes -0.0851375861
1353 NA12546 Yes -0.0950059013
1423 NA11920 Yes -0.0717307965
1451 NA12776 Yes -0.0865534231
13291 NA07435 Yes -0.104468725
上面数据中,个体的PRS为正值,说明风险高,为负值,说明风险低。
正文
数据使用GWAS分析教程中的数据。
HapMap_3_r3_1.bed HapMap_3_r3_1.bim HapMap_3_r3_1.fam
1. 将数据转为plink文本文件
plink --bfile HapMap_3_r3_1 --recode --out a1
这里,共有165个样本,每个样本1457897个位点。

2. 对基因型数据进行质控
质控标准:
- geno 0.1 # SNP 缺失率大于10%
- maf 0.05 # maf大于0.05
- mind 0.1 # 样本缺失率大于10%
- hwe 1e-5 # 哈温平衡P值大于1e-5
质控命令:
plink --file a1 --mind 0.1 --geno 0.1 --hwe 1e-5 --maf 0.05 --recode --out b1
日志文件:
PLINK v1.90b6.26 64-bit (2 Apr 2022) www.cog-genomics.org/plink/1.9/
(C) 2005-2022 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to b1.log.
Options in effect:
--file a1
--geno 0.1
--hwe 1e-5
--maf 0.05
--mind 0.1
--out b1
--recode
1031523 MB RAM detected; reserving 515761 MB for main workspace.
.ped scan complete (for binary autoconversion).
Performing single-pass .bed write (1457897 variants, 165 people).
--file: b1-temporary.bed + b1-temporary.bim + b1-temporary.fam written.
1457897 variants loaded from .bim file.
165 people (80 males, 85 females) loaded from .fam.
112 phenotype values loaded from .fam.
0 people removed due to missing genotype data (--mind).
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 112 founders and 53 nonfounders present.
Calculating allele frequencies... done.
Warning: 225 het. haploid genotypes present (see b1.hh ); many commands treat
these as missing.
Total genotyping rate is 0.997378.
0 variants removed due to missing genotype data (--geno).
Warning: --hwe observation counts vary by more than 10%. Consider using
--geno, and/or applying different p-value thresholds to distinct subsets of
your data.
--hwe: 2 variants removed due to Hardy-Weinberg exact test.
344283 variants removed due to minor allele threshold(s)
(--maf/--max-maf/--mac/--max-mac).
1113612 variants and 165 people pass filters and QC.
Among remaining phenotypes, 56 are cases and 56 are controls. (53 phenotypes
are missing.)
--recode ped to b1.ped + b1.map ... done.
质控后的位点结果:样本数:165 位点数:1113612
其中表型数据中,56个为case,56个为control,53个表型数据缺失。
提取出表型数据:
awk '{print $1,$2,$6}' b1.ped >phe.txt
将数据去除缺失值,分为base和target两个数据集。
library(data.table)
library(tidyverse)
d1 = fread("phe.txt",na.strings = "-9")
head(d1)
dd = d1 %>% drop_na(V3)
set.seed(123)
re1 = dd %>% sample_n(42) %>% select(V1,V2)
head(re1)
head(dd)
re2 = dd %>% filter(!V2 %in% re1$V2) %>% select(V1,V2)
head(re2)
fwrite(re1,"name_target.txt",col.names = F,sep = " ",quote = F)
fwrite(re2,"name_base.txt",col.names = F,sep = " ",quote = F)
3. 提取base和target数据集
注意,正常来说,base和target应该避免有亲缘关系,应该是独立样本。这里没有检测独立性,分为两类,只为演示。
plink --file b1 --keep name_base.txt --recode --out base_dat
plink --file b1 --keep name_target.txt --recode --out target_dat
日志文件:
$ plink --file b1 --keep name_base.txt --recode --out base_dat
PLINK v1.90b6.26 64-bit (2 Apr 2022) www.cog-genomics.org/plink/1.9/
(C) 2005-2022 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to base_dat.log.
Options in effect:
--file b1
--keep name_base.txt
--out base_dat
--recode
1031523 MB RAM detected; reserving 515761 MB for main workspace.
.ped scan complete (for binary autoconversion).
Performing single-pass .bed write (1113612 variants, 165 people).
--file: base_dat-temporary.bed + base_dat-temporary.bim +
base_dat-temporary.fam written.
1113612 variants loaded from .bim file.
165 people (80 males, 85 females) loaded from .fam.
112 phenotype values loaded from .fam.
--keep: 70 people remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 70 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Warning: 75 het. haploid genotypes present (see base_dat.hh ); many commands
treat these as missing.
Total genotyping rate in remaining samples is 0.997139.
1113612 variants and 70 people pass filters and QC.
Among remaining phenotypes, 36 are cases and 34 are controls.
--recode ped to base_dat.ped + base_dat.map ... done.
$ plink --file b1 --keep name_target.txt --recode --out target_dat
PLINK v1.90b6.26 64-bit (2 Apr 2022) www.cog-genomics.org/plink/1.9/
(C) 2005-2022 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to target_dat.log.
Options in effect:
--file b1
--keep name_target.txt
--out target_dat
--recode
1031523 MB RAM detected; reserving 515761 MB for main workspace.
.ped scan complete (for binary autoconversion).
Performing single-pass .bed write (1113612 variants, 165 people).
--file: target_dat-temporary.bed + target_dat-temporary.bim +
target_dat-temporary.fam written.
1113612 variants loaded from .bim file.
165 people (80 males, 85 females) loaded from .fam.
112 phenotype values loaded from .fam.
--keep: 42 people remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 42 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Warning: 1 het. haploid genotype present (see target_dat.hh ); many commands
treat these as missing.
Total genotyping rate in remaining samples is 0.997773.
1113612 variants and 42 people pass filters and QC.
Among remaining phenotypes, 20 are cases and 22 are controls.
--recode ped to target_dat.ped + target_dat.map ... done.
4. 对base数据进行GWAS分析
这里,将性别作为协变量,将PCA的3个值作为协变量,进行GWAS分析,把表型数据单独提取出来。
「提取性别数据:」
awk '{print $1,$2,$5}' base_dat.ped >cov1.txt
结果:
$ head cov1.txt
1328 NA06989 2
1377 NA11891 1
1349 NA11843 1
1330 NA12341 2
1418 NA12275 2
13292 NA07051 1
1354 NA12400 2
1451 NA12777 1
1353 NA12383 2
1418 NA12273 2
「提取表型数据:」
awk '{print $1,$2,$6}' base_dat.ped >phe1.txt
结果文件:
$ head phe1.txt
1328 NA06989 2
1377 NA11891 2
1349 NA11843 1
1330 NA12341 2
1418 NA12275 1
13292 NA07051 2
1354 NA12400 2
1451 NA12777 2
1353 NA12383 2
1418 NA12273 1
「计算PCA结果」
plink --file base_dat --pca 3
结果文件:
$ head plink.eigenvec
1328 NA06989 -0.00375153 -0.143377 -0.335978
1377 NA11891 0.00185169 -0.0483789 -0.168089
1349 NA11843 -0.0239135 0.0350353 0.0549738
1330 NA12341 -0.0333797 -0.0264783 0.0933901
1418 NA12275 -0.0184984 -0.156233 0.121645
13292 NA07051 -0.0219551 -0.0393832 -0.0171629
1354 NA12400 -0.0358468 -0.103815 -0.0732348
1451 NA12777 -0.0237697 -0.0013729 0.125297
1353 NA12383 -0.0167586 0.050416 -0.0170558
1418 NA12273 -0.0388465 -0.00396318 -0.204234
「将性别和PCA合并为协变量:」
awk '{print $3,$4,$5}' plink.eigenvec >pca.txt
paste cov1.txt pca.txt |sed 's/\s\+/ /g' >covar.txt
结果文件:
$ head covar.txt
1328 NA06989 2 -0.00375153 -0.143377 -0.335978
1377 NA11891 1 0.00185169 -0.0483789 -0.168089
1349 NA11843 1 -0.0239135 0.0350353 0.0549738
1330 NA12341 2 -0.0333797 -0.0264783 0.0933901
1418 NA12275 2 -0.0184984 -0.156233 0.121645
13292 NA07051 1 -0.0219551 -0.0393832 -0.0171629
1354 NA12400 2 -0.0358468 -0.103815 -0.0732348
1451 NA12777 1 -0.0237697 -0.0013729 0.125297
1353 NA12383 2 -0.0167586 0.050416 -0.0170558
1418 NA12273 2 -0.0388465 -0.00396318 -0.204234
进行logistic模型分析:
注意,添加--hide-covar,去掉协变量信息。
plink --file base_dat --pheno phe1.txt --logistic --covar covar.txt --out re1 --hide-covar
结果文件:
$ head re1.assoc.logistic
CHR SNP BP A1 TEST NMISS OR STAT P
1 rs3131972 742584 A ADD 70 2.484 1.712 0.08697
1 rs3131969 744045 A ADD 70 4.38 2.429 0.01513
1 rs1048488 750775 C ADD 69 2.646 1.811 0.07011
1 rs12562034 758311 A ADD 70 0.9018 -0.1598 0.8731
1 rs12124819 766409 G ADD 70 0.9347 -0.1684 0.8663
1 rs4040617 769185 G ADD 70 3.977 2.277 0.02278
1 rs4970383 828418 A ADD 70 1.974 1.238 0.2158
1 rs4475691 836671 T ADD 70 1.474 0.6384 0.5232
1 rs1806509 843817 C ADD 70 2.549 2.247 0.02465
「概率和对数优势比的关系」
进而可以推断出:
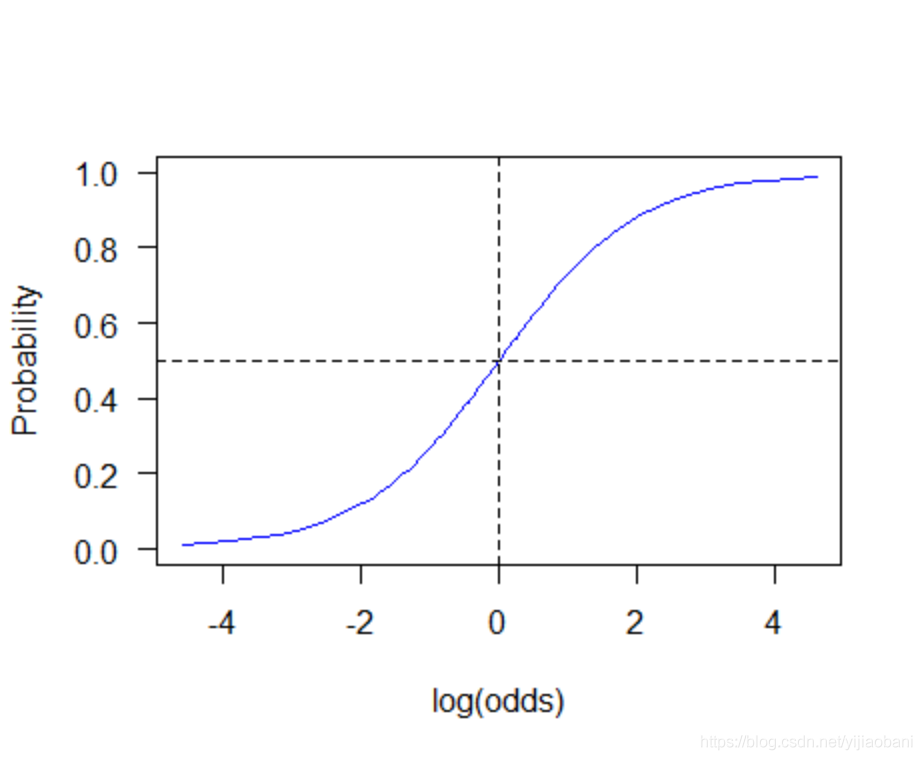
由图可知,概率P的最小值为0,最大值为1,中间值为0.5, 它对应的对数优势比分别是无穷小,无穷大和0.即:
- P=0, log(odds) = -Inf(负无穷大)
- P = 0.5, log(odds) = 0
- P = 1, log(odds) = Inf(正无穷大)
因此,Logistic回归常常用于估计给定暴露水平时结果事件发生的概率。例如,我们可以用它来预测在给定年龄、性别和行为方式等情形下某人患病的概率。
5. target计算PRS
这里,将target,分别提取性别和pca信息,表型数据,并将ped中的表型数据定义为-9(缺失)。
awk '{print $1,$2,$5}' target_dat.ped >cov1.txt
awk '{print $1,$2,$6}' target_dat.ped >phe1.txt
plink --file target_dat --pca 3
awk '{print $3,$4,$5}' plink.eigenvec >pca.txt
paste cov1.txt pca.txt |sed 's/\s\+/ /g' >covar.txt
awk '{$6=-9;print $0}' target_dat.ped >t.ped
mv t.ped target_dat.ped
这里的target_dat,为没有表型数据的文件。
将其转为二进制文件:
plink --file target_dat --make-bed --out target_bi
计算PRS:
Rscript PRSice.R --dir re1 --prsice ./PRSice_linux --base re1.assoc.logistic --target target_bi --thread 1 --stat OR --binary-target T --pheno phe1.txt
注意,phe1.txt,有无行头名都可以,都能够正常执行。上面计算PRS时也可以加入协变量,这里不再展示。
5. PRS结果说明
最适合的SNP个数是133个,R2位0.232258,P值为0.014
$ head PRSice.summary
Phenotype Set Threshold PRS.R2 Full.R2 Null.R2 Prevalence Coefficient Standard.Error P Num_SNP
- Base 0.00115005 0.232258 0.232258 0 - 17.1644 6.99987 0.0142024 133
对应的43个个体的PRS结果:
$ head PRSice.best
FID IID In_Regression PRS
1328 NA06984 Yes 0.0386390029
13291 NA06986 Yes -0.136642922
1418 NA12272 Yes -0.140312974
1421 NA12287 Yes -0.154526751
1330 NA12340 Yes -0.0851375861
1353 NA12546 Yes -0.0950059013
1423 NA11920 Yes -0.0717307965
1451 NA12776 Yes -0.0865534231
13291 NA07435 Yes -0.104468725
上面数据中,个体的PRS为正值,说明风险高,为负值,说明风险低。
结果可视化:


本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-10-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号