转录组差异分析FPKM与count处理差别大吗


缘起
这些天来,我们一般都是处理上游定量好的count数据,然后进行下游的转录组分析。但是,我们查看GEO数据集时,会发现有些数据集并没有提供count数据,而仅仅提供了FPKM或者RPKM等格式的数据。那当数据集提供的是FPKM数据集时,我们还能处理吗。前面曾老师分享的推文中描述了FPKM的处理方式,具体见RNAseq数据,下载GEO中的FPKM文件后该怎么下游分析,评论区中有小伙伴谈到limma包的作者不推荐用limma处理FPKM数据,最好用原始数据进行分析。那用count与用FPKM去处理获得的差异基因具有巨大的差别吗?曾老师前两天提出了这个疑问,于是便有了今天的推文。接下来,我们就探索一下用count与用FPKM去处理获得的差异基因是否具有巨大差别吧?
探究
今天,我们使用标题为 LncRNA-directed Antigenicity Loss Suppresses Immunosurveillance 的数据集 GSE113143 进行探究,数据集的介绍链接如下https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE113143 ,感兴趣的小伙伴可以点进去看看作者的总体设计。在此,我们对文章进行简单归纳,作者通过转录组测序探究了小鼠乳腺癌组织和乳腺组织的表达谱。。
转录组数据集介绍
GSE113143 数据集的样本分组如下,两个分组三个重复样本:

处理数据的话,作者仅仅提供了「FPKM矩阵」。因为我们要比较FPKM与count分别进行差异分析的区别,所以我们需要自身对于上游转录组数据进行定量,去获得「Ensenmble count矩阵」。之后,我们就可以直接走Ensenmble矩阵的差异分析流程进行分析了,链接见https://mp.weixin.qq.com/s/BHt_7WFCtKXIZwQYqhO8jA。
以下为作者提供的FPKM矩阵下载链接:https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE113143&format=file&file=GSE113143%5FNormal%5FTumor%5FExpression%2Etab%2Egz;感兴趣的小伙伴可以下载试试。
正式分析
1.利用fpkm值进行差异分析
FPKM处理代码援引自泥人吴老师的RNAseq数据,下载GEO中的FPKM文件后该怎么下游分析,其中也有一些很好的描述,感兴趣的小伙伴可以看看。FPKM处理关键是要将FPKM值转换为TPM值,然后用Limma进行后续的差异分析。
# 1.下载GSE113143数据集
a=read.table('GSE113143_Normal_Tumor_Expression.tab.gz',sep='\t',quote = "",fill = T,
comment.char = "!",header = T) # 提取表达矩阵
rownames(a)=a[,1]
a <- a[,-1]
# 2.将FPKM值转换为TPM值
expMatrix <- a
fpkmToTpm <- function(fpkm)
{
exp(log(fpkm) - log(sum(fpkm)) + log(1e6))
}
tpms <- apply(expMatrix,2,fpkmToTpm) #每列的基因
tpms[1:3,]
## N1 N2 N3 T1 T2 T3
## 0610005C13Rik 0.2320471 0.1715468 0.00000 0.00000 0.00000 0.00000
## 0610007P14Rik 48.3908046 39.2631560 46.04003 50.03695 59.05140 67.28521
## 0610009B22Rik 47.4905907 58.5953867 54.26887 49.78854 53.12592 57.99844
colSums(tpms) #都是每一百万(深度一样)
## N1 N2 N3 T1 T2 T3
## 1e+06 1e+06 1e+06 1e+06 1e+06 1e+06
# 3.差异分析
group_list=c(rep('Normal',3),rep('Tumor',3))
## 强制限定顺序
group_list <- factor(group_list,levels = c("Normal","Tumor"),ordered = F)
#表达矩阵数据校正
exprSet <- tpms
boxplot(exprSet,outline=FALSE, notch=T,col=group_list, las=2)
library(limma)
exprSet=normalizeBetweenArrays(exprSet)
boxplot(exprSet,outline=FALSE, notch=T,col=group_list, las=2)
#判断数据是否需要转换
exprSet <- log2(exprSet+1)
#差异分析:
dat <- exprSet
design=model.matrix(~factor( group_list ))
fit=lmFit(dat,design)
fit=eBayes(fit)
options(digits = 4)
topTable(fit,coef=2,adjust='BH')
## logFC AveExpr t P.Value adj.P.Val B
## Gm2897 -1.1291 0.5645 -314.35 1.062e-10 2.593e-06 10.205
## Tceal3 -2.4753 1.2376 -168.28 1.638e-09 2.000e-05 9.958
## Frk 1.9861 3.1418 114.45 8.853e-09 6.269e-05 9.600
## Nek2 2.2325 3.9233 108.20 1.132e-08 6.269e-05 9.527
## Calca -8.6440 4.3220 -105.14 1.283e-08 6.269e-05 9.487
## Paqr9 -4.1108 2.5527 -90.86 2.431e-08 9.368e-05 9.257
## Rpl39l -2.0242 1.0121 -88.82 2.685e-08 9.368e-05 9.217
## Fads3 -1.6945 5.7675 -80.69 4.086e-08 1.247e-04 9.033
## 4930578C19Rik -1.4385 0.7193 -77.15 4.974e-08 1.350e-04 8.939
## Smpd1 -0.8319 5.0280 -74.19 5.902e-08 1.441e-04 8.853
bp=function(g){
library(ggpubr)
df=data.frame(gene=g,stage=group_list)
p <- ggboxplot(df, x = "stage", y = "gene",
color = "stage", palette = "jco",
add = "jitter")
# Add p-value
p + stat_compare_means()
}
deg=topTable(fit,coef=2,adjust='BH',number = Inf)
head(deg)
## logFC AveExpr t P.Value adj.P.Val B
## Gm2897 -1.129 0.5645 -314.35 1.062e-10 2.593e-06 10.205
## Tceal3 -2.475 1.2376 -168.28 1.638e-09 2.000e-05 9.958
## Frk 1.986 3.1418 114.45 8.853e-09 6.269e-05 9.600
## Nek2 2.232 3.9233 108.20 1.132e-08 6.269e-05 9.527
## Calca -8.644 4.3220 -105.14 1.283e-08 6.269e-05 9.487
## Paqr9 -4.111 2.5527 -90.86 2.431e-08 9.368e-05 9.257
#save(deg,file = 'deg.Rdata')
# 4.FPKM转换为TPM后,再利用limma进行差异分析
## 不同的阈值,筛选到的差异基因数量就不一样,后面的超几何分布检验结果就大相径庭。
if(T){
logFC_t=1.5
deg$g=ifelse(deg$P.Value>0.05,'stable',
ifelse( deg$logFC > logFC_t,'UP',
ifelse( deg$logFC < -logFC_t,'DOWN','stable') )
)
table(deg$g)
head(deg)
}
## logFC AveExpr t P.Value adj.P.Val B g
## Gm2897 -1.129 0.5645 -314.35 1.062e-10 2.593e-06 10.205 stable
## Tceal3 -2.475 1.2376 -168.28 1.638e-09 2.000e-05 9.958 DOWN
## Frk 1.986 3.1418 114.45 8.853e-09 6.269e-05 9.600 UP
## Nek2 2.232 3.9233 108.20 1.132e-08 6.269e-05 9.527 UP
## Calca -8.644 4.3220 -105.14 1.283e-08 6.269e-05 9.487 DOWN
## Paqr9 -4.111 2.5527 -90.86 2.431e-08 9.368e-05 9.257 DOWN
fpkm_deg=deg
2.利用count值进行差异分析
# 自身走上游定量,获得count-ensenmble矩阵
library(edgeR)
library(DESeq2)
library(FactoMineR)
library(factoextra)
library(clusterProfiler)
library(org.Mm.eg.db)
library(stringr)
library(stringi)
library(tidyverse)
library(ggplot2)
library(patchwork)
library(pheatmap)
library(VennDiagram)
library(RColorBrewer)
library(patchwork)
library(ggplotify)
# 1.读取数据,获得基因表达矩阵以及cpm矩阵
rawcount <- read.table("./all.id.txt",header = T,sep="\t",row.names = 1)
colnames(rawcount)
## [1] "Chr" "Start"
## [3] "End" "Strand"
## [5] "Length" "...3.hisat2.SRR7003811.sort.bam"
## [7] "...3.hisat2.SRR7003812.sort.bam" "...3.hisat2.SRR7003813.sort.bam"
## [9] "...3.hisat2.SRR7003814.sort.bam" "...3.hisat2.SRR7003815.sort.bam"
## [11] "...3.hisat2.SRR7003816.sort.bam"
rawcount <- rawcount[,6:11]
##转换ID
id2symbol <- bitr(rownames(rawcount),
fromType = "ENSEMBL",
toType = "SYMBOL",
OrgDb = org.Mm.eg.db)
rawcount <- cbind(GeneID=rownames(rawcount),rawcount) #多一列换成基因ID
rawcount <- merge(id2symbol,rawcount,
by.x="ENSEMBL",by.y="GeneID",all.y=T)
rawcount=rawcount[!duplicated(rawcount$SYMBOL),]
rawcount <- drop_na(rawcount) #去除含有NA值的行
rawcount <- rawcount[,!colnames(rawcount)%in%c("ENSEMBL","Ensmble","median")]#正式获得基因rawcount矩阵
keep <- rowSums(rawcount>0) >= floor(0.5*ncol(rawcount))
filter_count <- rawcount[keep,] #获得filter_count矩阵
rownames(filter_count) <- filter_count$SYMBOL
filter_count <- filter_count[,!colnames(filter_count)%in%"SYMBOL"]
express_cpm <- log2(cpm(filter_count)+ 1)
express_cpm[1:4,1:4] #获得cpm矩阵
## ...3.hisat2.SRR7003811.sort.bam ...3.hisat2.SRR7003812.sort.bam
## Gnai3 7.152 7.100
## Cdc45 3.665 3.190
## H19 3.732 7.790
## Scml2 1.572 1.546
## ...3.hisat2.SRR7003813.sort.bam ...3.hisat2.SRR7003814.sort.bam
## Gnai3 7.235 7.684
## Cdc45 3.345 4.736
## H19 5.189 5.999
## Scml2 1.731 1.485
# 3.用limma进行差异分析
#根据列的信息,提取分组信息
colnames(rawcount) =c(rep('Normal',3),rep('Tumor',3))
group=rep(c("WT","Tumor"),each=3)
group_list=factor(group,levels = c("WT","Tumor"))
table(group_list)#检查一下组别数量
## group_list
## WT Tumor
## 3 3
# 4.limma----voom差异分析
library(limma)
exp=filter_count
Group=group_list
dge <- edgeR::DGEList(counts=exp)
dge <- edgeR::calcNormFactors(dge)
design <- model.matrix(~Group)
v <- voom(dge,design, normalize="quantile")
fit <- lmFit(v, design)
fit= eBayes(fit)
DEG_limma = topTable(fit, coef=2, n=Inf)
DEG_limma = na.omit(DEG_limma)
pvalue_t=0.05
logFC_t=1.5
k1 = (DEG_limma$P.Value < pvalue_t)&(DEG_limma$logFC < -logFC_t)
k2 = (DEG_limma$P.Value < pvalue_t)&(DEG_limma$logFC > logFC_t)
DEG_limma$regulated = ifelse(k1,"down",ifelse(k2,"up","not"))
table(DEG_limma$regulated)
##
## down not up
## 2230 15771 1709
3.比较分别用count与fpkm进行差异分析的结果
## 001 绘制整体基因的相关性散点图
ids=intersect(rownames(fpkm_deg),rownames(DEG_limma))
# 01看一看分别比较的整体相关性(此处就看整体差异基因吧)
library(ggstatsplot)
library(ggpmisc)
# 02构建可视化所需的矩阵(相关性就两列值)
df=data.frame(
deg1=fpkm_deg[ids,"logFC"],
deg2=DEG_limma[ids,"logFC"] #就是两列logFC的值(散点图)
)
# 03绘制相关性图形
p <- ggplot(df, aes(x=df$deg1,
df$deg2))+
geom_point()+
labs(x = "all_fpkm",
y = "all_count")+
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept = c( -1.5,0,1.5),lty=4,col="#ec183b",lwd=0.8) +
geom_hline(yintercept = c( -1.5,0,1.5),lty=4,col="#18a5ec",lwd=0.8) +
#xlim(-5,5)+
#ylim(-3,3)+
theme_ggstatsplot()+
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_blank())
p+stat_poly_eq(aes(label=paste(..eq.label..,..adj.rr.label..,..p.value.label..,sep = "~~~~")),formula = y~x,parse=T,size=3.0)
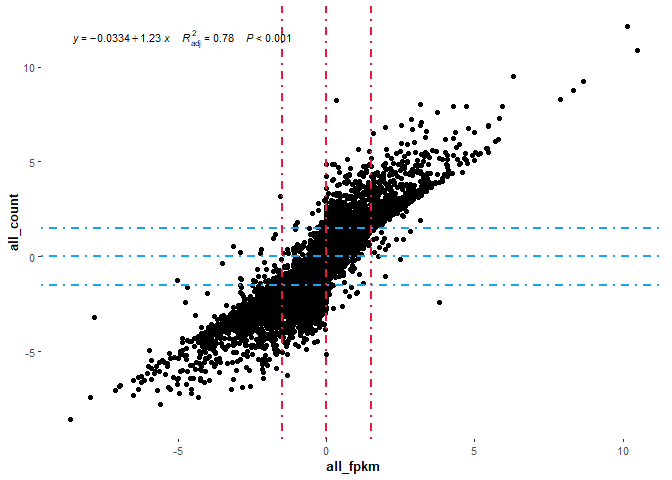
## 002 再绘制差异基因的相关性散点图看看(整体都没有问题,差异肯定没有问题)
deg_limma=rownames(DEG_limma[DEG_limma$regulated!="not",])
deg_fpkm=rownames(fpkm_deg[fpkm_deg$g!="stable",])
ids=intersect(deg_limma,deg_fpkm)
# 01看一看分别比较的整体相关性(此处就看整体差异基因吧)
library(ggstatsplot)
library(ggpmisc)
# 02构建可视化所需的矩阵(相关性就两列值)
df=data.frame(
deg1=fpkm_deg[ids,"logFC"],
deg2=DEG_limma[ids,"logFC"] #就是两列logFC的值(散点图)
)
# 03绘制相关性图形
p <- ggplot(df, aes(x=df$deg1,
df$deg2))+
geom_point()+
labs(x = "deg_fpkm",
y = "deg_count")+
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept = c( -1.5,0,1.5),lty=4,col="#ec183b",lwd=0.8) +
geom_hline(yintercept = c( -1.5,0,1.5),lty=4,col="#18a5ec",lwd=0.8) +
#xlim(-5,5)+
#ylim(-3,3)+
theme_ggstatsplot()+
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_blank())
p+stat_poly_eq(aes(label=paste(..eq.label..,..adj.rr.label..,..p.value.label..,sep = "~~~~")),formula = y~x,parse=T,size=3.0)
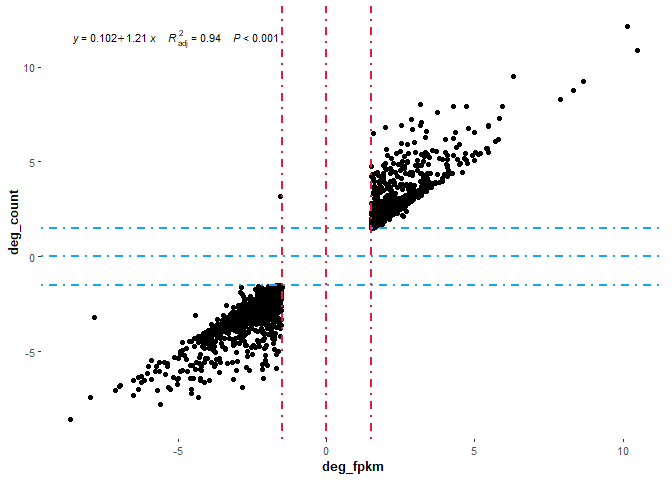
以上演示了count与FPKM进行差异分析的方式,并通过散点图展示了两者差异分析后获得的差异基因的区别,两种方式都是用limma包进行差异分析,感兴趣的小伙伴们也可以尝试下哈。
两个散点图分别分析了FPKM与count进行差异分析后整体差异基因的相关性与差异基因的相关性。从图中,我们可以看出FPKM的差异分析结果与count的差异分析结果基本一致,可能算法在数据的差异中并非起绝对作用。但为什么limma包不提倡用FPKM以及有的推文说用FPKM错了呢,然而此处针对FPKM与count进行差异分析,为什么结果没有很明显的区别呢?小编觉得是一个很好的问题,希望明白的小伙伴可以补充下。
如果有小伙伴或者很厉害的大牛觉得我的观点有问题有不妥,欢迎投稿给曾老师。小编一定给您排好版,将您的知识传递给小伙伴们,让大家更好的交流。小编自身也是个小菜鸟,受曾老师感染,在此处更多想做的就是传递自己的认知,分享自身的代码,写好这部分内容。希望迎来的不是很霸气的没有义务,劝小编退更的话语,谢谢。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-12-14,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号