泛基因组文献007~Genome Biology 玉米泛基因组
泛基因组文献007~Genome Biology 玉米泛基因组

论文
A pan-Zea genome map for enhancing maize improvement
https://link.springer.com/article/10.1186/s13059-022-02742-7#availability-of-data-and-materials
s13059-022-02742-7.pdf
提供了数据处理流程
https://github.com/songtaogui/pan-Zea_construct/tree/v1.0.0
仔细看看论文,然后试着这个流程
首先是流程的安装
整个流程是shell写的,依赖软件
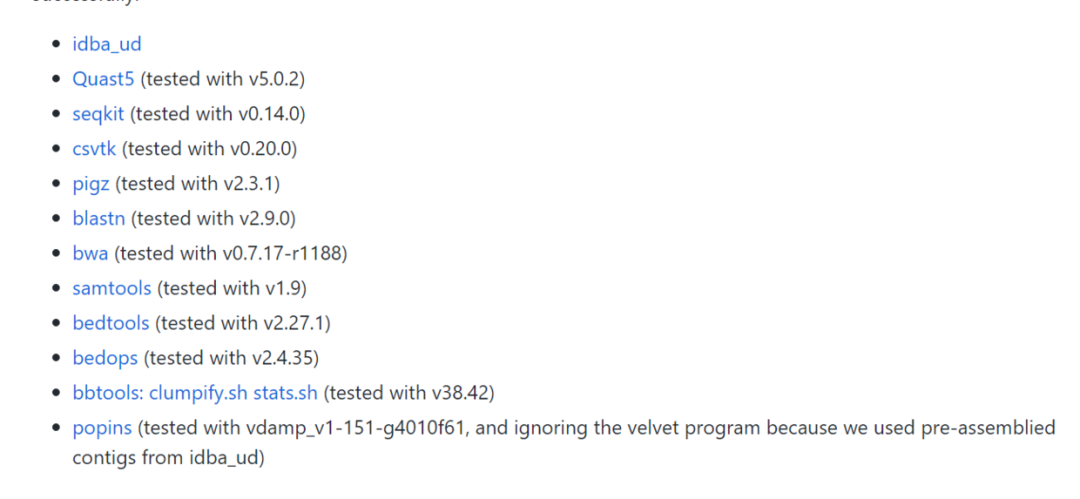
image.png
大部分都可以用conda安装
- bbtools这个软件 conda安装的时候 是安装 bbmaps
- popins这个软件是不能用conda装的
github主页 https://github.com/bkehr/popins
这个软件也有一些依赖的软件,大部分都可以用conda安装,但是那个seqan好像不行,seqan的帮助文档
https://seqan.readthedocs.io/en/master/Infrastructure/Use/Install.html#infra-use-install
安装需要用到root,课题组服务器我是没有root权限的,我就用我阿里云的服务器来安装
apt install libseqan2-dev
安装在目录 /usr/include/ 下
把seqan这个文件夹放到课题组服务器上
和popins放到一个目录下
然后再popins目录下运行 make
安装成功了,多了一个popins的可执行文件
这里运行pan04_popins_pipe.sh 是使用popins这个软件,这里会有报错会有报错,查了一下这个链接有讨论,这个链接的讨论是玉米泛基因组论文的作者
https://github.com/bkehr/popins/issues/4
这里seqan需要是2.2版本 下载链接
https://github.com/seqan/seqan/releases/tag/seqan-v2.2.0
安装命令
sudo apt install ./seqan-library-2.2.0.deb
重新安装popins
make clean
make
- quast用conda也一直没有安装成功 ,可能是和其他软件的依赖软件有冲突
我在另外的conda环境中安装过quast,我用软连接把之前的安装可执行文件链接到当前的conda环境下
ln -s /mnt/shared/scratch/myan/apps/mingyan/Biotools/mambaforge/envs/genome_assembly/bin/quast /mnt/shared/scratch/myan/apps/mingyan/Biotools/mambaforge/envs/pan_zea/bin/
quast 需要把quast.py链接过来
把popins也用软连接链接到这个环境下
ln -s /mnt/shared/scratch/myan/private/pome_WGS/pan_zea/popins/popins /mnt/shared/scratch/myan/apps/mingyan/Biotools/mambaforge/envs/pan_zea/bin/
这样能行吗,不太确定,目前查看帮助文档内容反正是没有问题
运行命令
blastdbcmd -db /mnt/shared/apps/databases/ncbi/nt -entry all -outfmt "%g,%l,%T" > nt_all_accession_length_taxid.csv
遇到报错
Error: [blastdbcmd] error while reading seqid
把blast从2.9更新到2.13就可以了
运行整个流程
PANZ_individual_pipe.sh
运行完第一步就会有报错,shell的内容大体能看懂,大概的错误就是因为路径的问题找不到第二步的输入文件,但是具体怎么改还是没有想明白,尝试着把每一步单独运行
bash pan-Zea_construct/src/pan00_IDBA_assembly.sh SRR1946554 01.clean.fq/SRR1946554_clean_1.fastq.gz 01.clean.fq/SRR1946554_clean_2.fastq.gz 500 8
bash pan-Zea_construct/src/pan01_quast_pre-unaln.sh SRR1945464 /home/myan/scratch/private/pome_WGS/pan_zea/Arabidopsis/SRR1945464/SRR1945464_idba/SRR1945464_scaftig500.fa.gz /home/myan/scratch/private/pome_WGS/pan_zea/Arabidopsis/ref/at.fa 150 500 8
bash pan-Zea_construct/src/pan02_blastNT_clean.sh SRR1945464 SRR1945464.unaligned.150bp.fa.gz SRR1945464/SRR1945464_idba/SRR1945464_scaftig500.fa.gz SRR1945464_quast/contigs_reports/contigs_report_SRR1945464_scaftig500.unaligned.info 8 /mnt/shared/apps/databases/ncbi/nt plant_nt_accession.txt 0.4 100 500
bash pan-Zea_construct/src/pan03_reMEM_filter.sh SRR1945464 SRR1945464_unrefseq_kept.fa.gz ref/at.fa 0.8 0.9 12
bash pan-Zea_construct/src/pan04_popins_pipe.sh SRR1945464 03.sorted.bam/SRR1945464.sorted.bam SRR1945464.unaligned.MEMfiltered.fa.gz ref/at.fa 101 100 16
bash pan-Zea_construct/src/pan05_pmrc_filter.sh SRR1945464 SRR1945464.unaligned.MEMfiltered.fa.gz pp_SRR1945464/SRR1945464/non_ref_new.bam 10 8
bash pan-Zea_construct/src/pan06_get_mmanchor.sh SRR1945464 SRR1945464.unaligned.pmrcfiltered.fa.gz SRR1945464_quast/contigs_reports/minimap_output/SRR1945464_scaftig500.coords.filtered 8
bash pan-Zea_construct/src/pan07_get_pploc_qrg_rd.bash SRR1945464 pp_SRR1945464/clean_nonrefseq_4_SRR1945464_locations.txt pp_SRR1945464/SRR1945464/non_ref_new.bam 10 8
bash pan-Zea_construct/src/pan08_combine_mm_pp.sh SRR1945464 SRR1945464_mmanchor_fmt.bed SRR1945464_pploc_qrg_rd_fmt.tsv 8
bash pan-Zea_construct/src/pan09_MMPPSR.sh SRR1945464 3 3 SRR1945464/SRR1945464_vcf_header.txt pp_SRR1945464/clean_nonrefseq_4_SRR1945464_insertions.vcf SRR1945464_anchor_combine_mm_pp.tsv 8
其中有一步用到samtools会遇到报错 samtools sort: couldn't allocate memory for bam_mem这一步不要用太多核心,增加需要的内存,(我最开始用了16个G,换到64G后就没有报错了)
这里相当于是运行了一个样本,第一步idba和第三步blast比对NT数据库需要的时间很长
我这里用到的数据是拟南芥的数据
所有样本运行完 还要运行 PANZ_cluster_pipe.sh,等运行完所有样本再来记录这个过程