Drug Discov Today|化学分子指纹的概念和应用


2022年9月13日,哈尔滨医科大学药物基因组信息学教研室陈秀杰教授、哈尔滨医科大学生物信息科学与技术学院解洪波副教授团队在期刊Drug Discovery Today上发表论文“Concepts and applications of chemical fingerprint for hit and lead screening”。

论文中,作者总结了8类分子指纹,并介绍了分子指纹在虚拟筛选等8种下游任务中的应用。本文对计算药物研发选择合适的分子指纹提供了良好的总结和指南。
1 摘要
分子指纹(Molecular fingerprints)可以低计算成本的方式表示大规模化学数据集中化合物的化学(结构、物理化学等)性质。它们在将化学数据集中的分子转换为适合于计算方法的一致输入格式(bit向量或数值)方面发挥着重要作用。在这篇综述中,作者将常见和最先进的分子指纹归纳并分类为8种不同类型(基于字典的、圆形的(circular)、拓扑的、药效团的(pharmacophore)、蛋白质-配体相互作用的、基于形状的、强化的和多种的)。作者还强调了分子指纹在早期药物研发中的应用。因此,本综述为药物研发使用合适的化合物(或配体-蛋白质复合物)指纹的选择提供了指南。
2 介绍
化学指纹或描述符最初用于表示分子的结构特征,在计算方法中,它是将分子结构与物理化学性质和生物活性联系起来的桥梁。随着化学、统计学和计算机科学等学科之间的壁垒被打破,各种类型的分子表示方法应运而生。在构效关系研究中,分子的子结构单元或物理化学性质被称为“描述符”。化学结构表示可以捕捉从0D到4D的不同性质的化合物。随着对快速子结构搜索和机理分析的需求日益增长,代表化学分子局部结构特征和物理化学性质的指纹已迅速发展。
指纹(或描述符)可以提取适合作为机器学习和QSAR模型输入的结构信息以及物理化学性质和生物活性,是能将化学分子转换为一致形式的数字或向量表示的一种有力工具。一个通用的分子指纹或描述符通常具有以下特征:
1.能够表示分子的局部结构(即能够表示每个原子及其最近邻居);
2.能够组合并有效且简单地表示分子结构或物理化学性质;
3.能够从编码的指纹中高效、简单地解码为分子结构;
4.分子指纹中的特征是相互独立的。
3 分子指纹的类型
指纹算法简单而快速地将化学结构或性质转换为高度压缩的表示,这有效地加速了虚拟筛选过程。化学表示的选择直接影响高通量筛选(high-throughput screening, HTS)方法的准确性和应用范围。不同类型的指纹在不同的要求和背景数据集下具有不同的性能。作者对常见分子指纹进行了分类,并简要总结了每种分子指纹能够捕获的结构和物理化学特征,并为如何根据特定研究要求选择化合物(或配体-蛋白质复合物)的合理分子指纹表示提供了建议。传统和最先进分子指纹的8种类型和应用概述如图1所示。
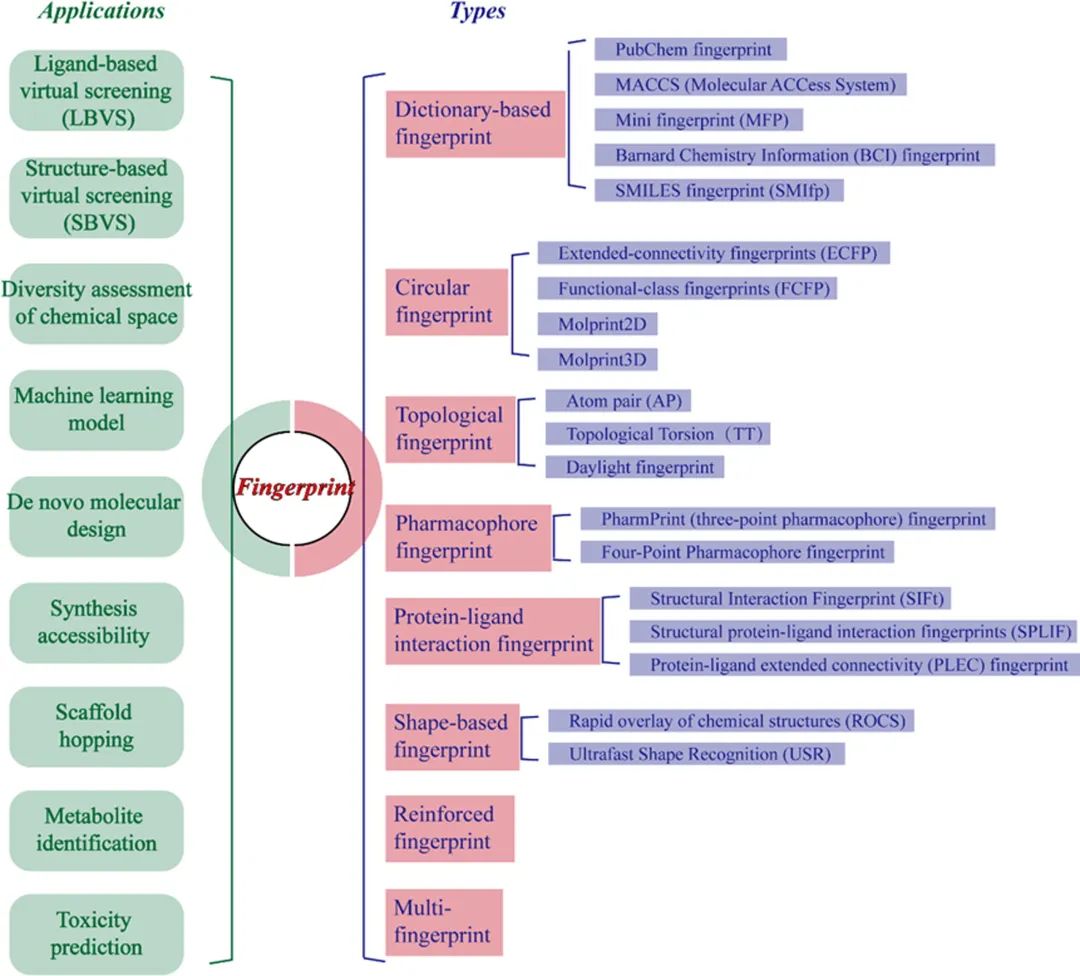
图1 分子指纹的常见类型和应用概览。
3.1 基于字典的分子指纹
基于字典的分子指纹(Dictionary-based (structural keys) fingerprints):表示为长度为的向量,每个位置为1或0,1表示该分子存在预定义的某种官能团(functional groups)、子结构基序(substructure motifs)、片段(fragments),0则表示该分子不存在预定义的这种官能团、子结构模体、片段。
常见的基于字典的指纹类型有:(1) PubChem (PC) fingerprints, (2) Molecular ACCess System (MACCS), (3) Mini FingerPrint (MFP), (4) Barnard Chemistry Information (BCI) fingerprints, (5) SMIles FingerPrint (SMIFP).
3.2 圆形分子指纹
圆形分子指纹(Circular fingerprints),早在1965年就以Morgan算法的形式首次提出。与基于字典的指纹不同,圆形指纹通常会捕获新的片段。圆形指纹产生单个、独立的结构片段,这些片段呈圆形。圆形指纹算法通常以分子中的每一个非氢原子或分子片段为中心,并根据其特定的预定义规则将分子片段迭代地扩展到其邻居,直到分子的所有片段都被枚举(或直到迭代次数达到自定义数量)。
常见的圆形指纹类型有:(1) extended connectivity fingerprints (ECFPs), (2) functional-class fingerprints (FCFPs), (3) Molprint2D and Molprint3D.
3.3 拓扑(基于路径)的分子指纹
拓扑(基于路径)的分子指纹:Topological (path-based) fingerprints. 化学拓扑性质来源于化学图,数学上表示为,其中,是节点(原子)集合,为边(化学键)集合。2D分子结构通常基于其拓扑性质表示,例如2D连接表,它是MOL和SDF的化合物格式的基本单元(图2)。化合物的常见拓扑性质包括:(1) 原子类型;(2) 每个非氢原子(邻接矩阵)的连接性(或度);(3) 每对原子的拓扑距离(距离矩阵);(4) 原子离心率(atom eccentricity);(4) 通过特定方法确定的化学键和原子的重量(weights of bonds and atoms by specified custom approaches)。化合物的片段(或子结构)和物理化学性质是分类和预测生物活性的重要基础。
常见的拓扑指纹类型有:(1) atom pairs (APs), (2) topological torsion (TT), (3) Daylight fingerprints.
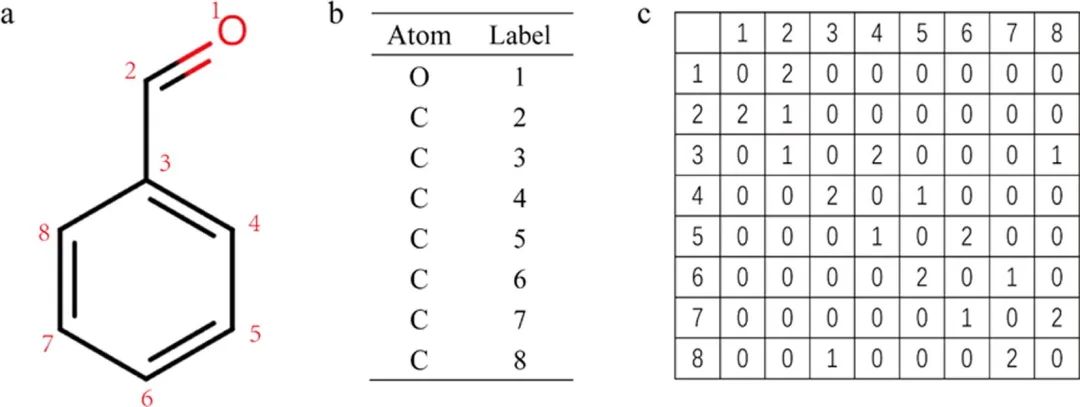
图2 以苯甲醛(benzaldehyde)为例,将化学分子转化为2D连接表。(a) 标记每个非氢的原子。(b) 标记二维连接表的第一列和第一行中的原子标签。(c) 苯甲醛的2D连接表。表中的数字表示原子之间的键类型。
3.4 药效团分子指纹
药效团分子指纹:Pharmacophore fingerprints. 在过去几十年,药效团建模一直是药物研发的一种关键和成功的方法,并且对分子表示和复杂生物系统分析具有重要影响。3D药效团是在3D空间中排列的药物-受体相互作用中观察到的一组化学或功能相互作用特征,如氢键、电荷转移、静电和疏水相互作用。这些特征代表有机配体与大分子受体的基本相互作用信息。基于药理学表示的分子指纹预期有助于表示配体-受体非共价结合的功能或特征。
常见的药效团指纹类型有:(1) PharmPrint (3-point PP), (2) 4-point PP.
3.5 蛋白质-配体相互作用分子指纹
蛋白质-配体相互作用分子指纹(Protein–ligand interaction fingerprints, PLIFP),用于通过分析和提取受体和配体之间的结合模式(或者是一组固定的氨基酸残基,称为基于残基的IFP,或者是原子的组合,称为基于原子的IFP)或物理化学特征来表示分子内相互作用。此类指纹可以使用关于分子对接或基于结构的实验数据的信息,将3D蛋白质-配体相互作用转换为1D bit串,随后用于比较蛋白质-配体的相互作用特异性。PLIFP中的这些位是通过特定规则计算的,例如原子类型和几何(距离或角度)测量。基于结合位点的相似性,可以推断出相互作用模式的相似性。这有助于从局部结构评估蛋白质-配体关系的结合模式。
常见的蛋白质-配体相互作用指纹类型有:(1) structural interaction fingerprints (SIFts), (2) structural protein–ligand interaction fingerprints (SPLIFs), (3) protein–ligand extended connectivity (PLEC) fingerprints.
3.6 基于形状的分子指纹
基于形状的分子指纹(Shape-based fingerprints),对于根据参考配体,进行基于形状和构象相似性的小分子虚拟筛选而言,是一种非常有效的特征。与其他虚拟筛选方法相比,基于形状的筛选在某些方面表现出更显著的性能,例如识别新的生物活性配体。基于形状的相似性评估通常指化学分子的空间体积和基于表面的形态、静电和药效特征等特性,其通常通过对齐方法(构建两个形状的3D覆盖)或通过数值比较特征向量方法(将形状减少为低维向量)来确定。
常见的基于形状的分子指纹有:(1) rapid overlay of chemical structures (ROCS), (2) ultrafast shape recognition (USR).
3.7 强化分子指纹
强化分子指纹:Reinforced fingerprints. 上文提到的6种经典分子指纹类型具有独特的算法、特征和应用,可以根据化学信息学的进步进行修改,从而形成强化分子指纹。作者在论文中总结了目前研究者提出的多种强化分子指纹。
3.8 多种分子指纹
多种分子指纹(Multi-fingerprints),即将多种分子指纹进行组合使用。单一的化学指纹不能够捕捉到化合物的所有关键结构或性质(或配体-靶相互作用)。然而,不同类型描述符的组合将捕获化合物的多种特征,这可以提高某些算法或模型中活动预测的性能。这种同时组合多个指纹的描述符的方法被称为多种分子指纹。例如,Lo等人提出的CSNAP3D结合了形状和药效团分子指纹特征,以测量配体之间的3D相似性,并在靶标预测任务上显示出显著的性能改进。
表1 不同分子指纹算法的特点
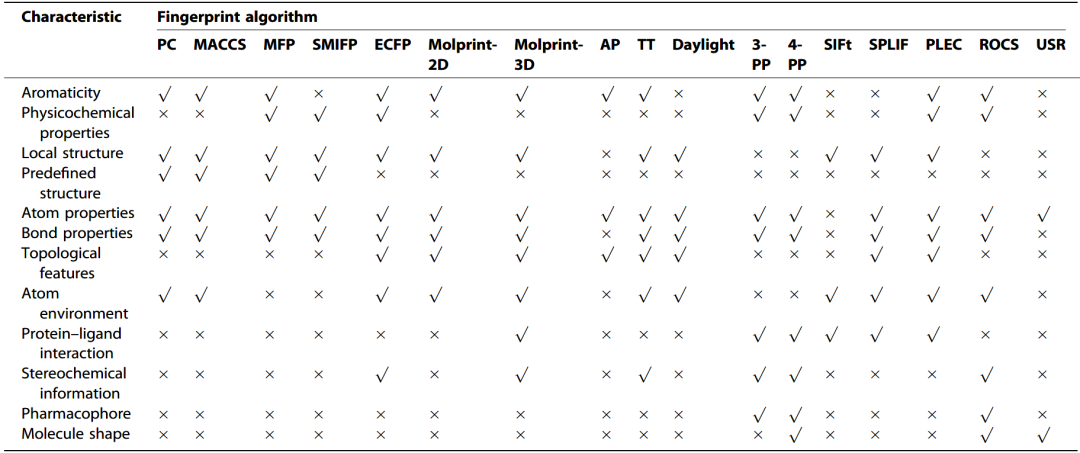
作者还在论文中总结了现有的多种分子指纹联合使用的研究进展。不同分子指纹算法的特点比较见表1。
4 应用
论文主要介绍了分子指纹8种类型的应用:(1) 虚拟筛选;(2) 化学空间的多样性评估;(3) 作为判别模型的特征;(4) 用于计算上的分子从头设计;(5) 合成可及性和反应预测(从反应物的分子指纹出发,应用神经网络进行反映预测的示例见图3);(6) 骨架跳跃(scaffold hopping);(7) 代谢产物识别;(8) 毒性预测。下面详细介绍分子指纹在高通量虚拟筛选种的应用。
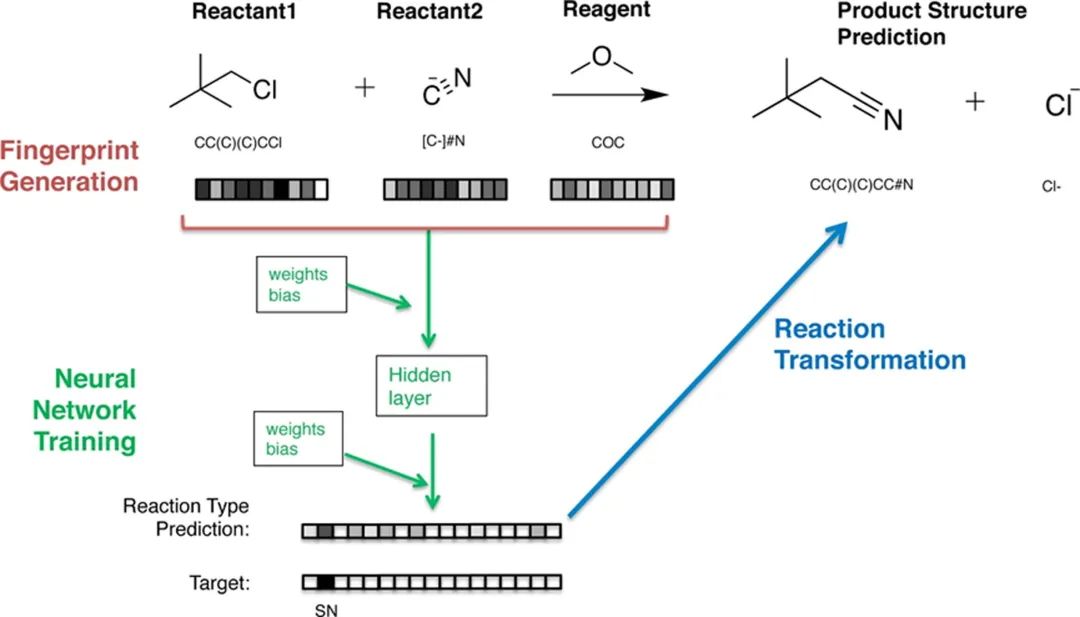
图3 通过反应指纹和神经网络预测反应类型和产物结构的过程。
4.1 虚拟筛选
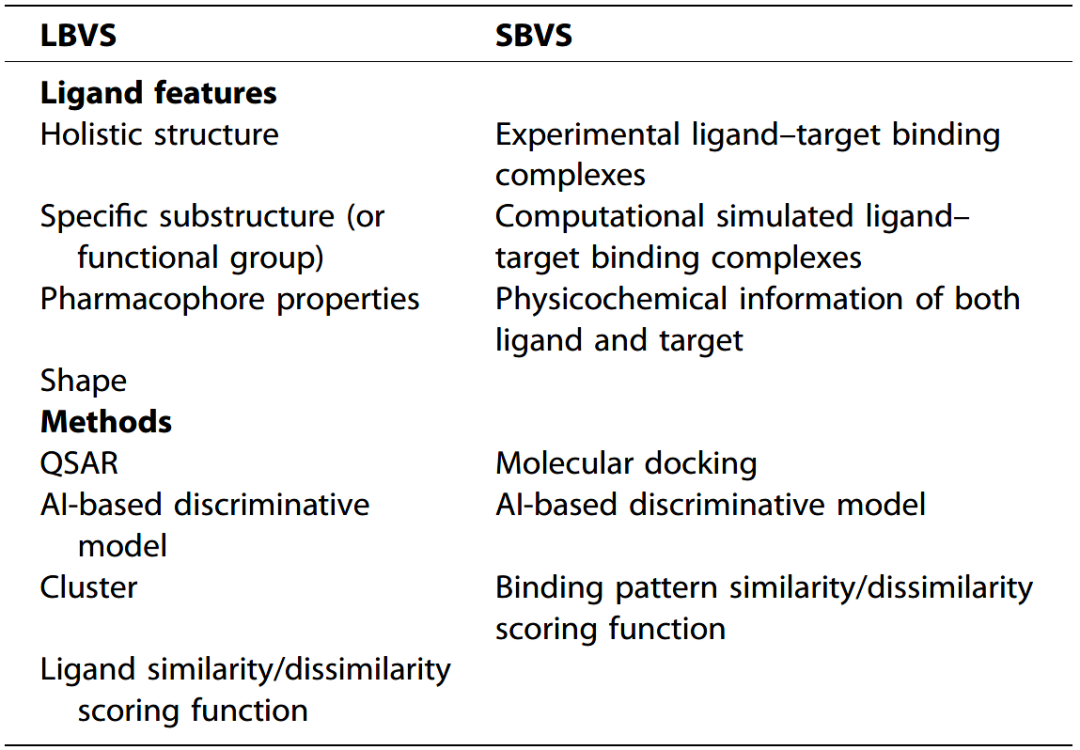
在早期药物研发过程中,最传统和最被接受的苗头化合物、先导化合物发现和优化的方法是基于一系列实验生物分析的高通量筛选(high-throughput screening, HTS)。鉴于实验方法的局限性,如成本高、持续时间长和效率低,虚拟筛选作为一种计算技术出现,可以自动从大规模数据集中识别有效的活性候选分子,以预先筛选出有潜力的化合物用于生物测试。虚拟筛选可分为两大类:基于配体的虚拟筛选(ligand-based virtual screening, LBVS)和基于结构的虚拟筛选(structure-based virtual screening, SBVS)。
LBVS基于结构相似的化合物具有相似生物活性的假设。其目的是识别与已知对特定靶标具有活性的化合物类似的化合物。相比之下,SBVS侧重于配体-靶标结合位点的结构信息和靶标蛋白质的3D结构知识,从化学空间中优先考虑和搜索对感兴趣靶标(蛋白质或RNA)有结合潜力的配体。两类虚拟筛选方法的比较见图2。
表2 LBVS和SBVS的比较
4.1.1 LBVS
LBVS通常只需要有关配体相关性质的信息,这具有效率高、时间短的优点。LBVS方法通常侧重于具有已知和未知活性的化合物之间的比较分析。比较分析主要基于化合物的四个方面:(1) 整体结构;(2) 特定子结构(或官能团);(3) 药效团性质[氢键供体(hydrogen bond donors, HBDs)、氢键受体(hydrogen bond acceptors, HBAs)等];(4) 分子3D形状。基于相似性原理(Similarity Property Principle, SPP)假设的相似性搜索是LBVS的典型方法。各种分子指纹或物理化学描述符来被用来识别与模板分子在结构或性质上的相似性/不相似性。结果取决于各种分子指纹方法以及它们如何计算化学分子的表示。例如,使用不同种类的分子指纹可以提取不同的配体性质。当使用结构性的分子指纹或描述符时,化学分子的结构信息和物理化学性质通常被表示向量或字符串,这是LBVS的关键步骤。化合物的密集、快速和计算机友好的表示有助于计算上的高通量虚拟筛选。
在大规模数据集中筛选特定的生物活性常用四种方法:(1) QSAR;(2) 基于AI的判别模型;(3) 聚类;(4) 相似性/不相似性评分函数的搜索方法。根据研究人员关注的特定需求或任务,不同的分子指纹或描述符将化学分子转换为矩阵,并通常作为QSAR和ML模型中的输入。
LBVS的相关方法主要基于配体本身的相似性比较。然而,由于“活性悬崖”,结构相似的化合物之间有时会出现生物活性的显著差异。仅依赖于配体的单一视角可能导致不准确虚拟筛选结果。
4.1.2 SBVS
SBVS主要基于配体-蛋白质复合物结合位点的结构,并且更全面,通常具有更大的数据量,从而提高了SBVS的性能和准确性。典型的基于结构的虚拟筛选过程通常包括两个步骤:(1) 基于形状和相互作用互补性的信息将化合物对接到推定的结合口袋(putative binding pocket);(2) 评估与靶标相互作用的化合物的特定构象的结合亲和力,并通过评分函数选择最有利的对接姿势。
自20世纪80年代初以来,分子对接一直是一种被广泛认可和有效的SBVS计算方法。其目的是将候选数据集中的所有配体对接到所选靶标的结合口袋中,然后评估配体与具有已知3D结构的蛋白质之间的亲和力。然而,用于测量配体-靶标亲和力的实验方法[如核磁共振(NMR)和X射线晶体学]耗时长使得分子对接不适合高通量虚拟筛选任务。因此,基于几何匹配和能量匹配的配体-蛋白质相互作用的计算机辅助自动建模的好处不言而喻。然而,在评估配体-蛋白质姿势的结合亲和力时,评分函数面临众所周知的局限性,例如对构象熵(例如,蛋白质柔性)和来自溶剂的能量贡献的考虑不足。蛋白质-配体相互作用指纹,例如IFP、SIFt和SPLIF,包含配体和靶标的结构和物理化学信息的蛋白质-配体复合物提供了一种快速和自动化的方式评估蛋白质-配体复合体的3D结构相似性的替代方法。
分析和评估配体-蛋白质结合相互作用的结构和姿势信息在SBVS中至关重要。传统的方法,如X射线晶体学、核磁共振和计算分子对接,在处理高通量任务时变得困难和低效。因此,基于分子指纹的机器学习算法允许简单和通用的结构解释和有效的数据处理。
5 总结和展望
分子指纹算法在简单性和表示性之间提供了良好的权衡,并且可以用于以低计算成本的方式提取和表达大规模化学集合的特定结构或物理化学特征。然而,尽管它们具有优势,但在药物研发应用中使用当前的分子指纹仍存在一些挑战。
1. 数据集质量低且可访问性差。公共数据库中,可用的信息和相关可用特征非常少,而许多制药公司每年也会产生大量未公开的实验数据。
2. 缺乏标准的一站式管道。尽管指纹具有各种优点,但它很少能够独立完成高通量虚拟筛选任务。在通过分子指纹制备化学分子的输入数据之后,需要将计算公式或预测模型作为任务的核心步骤,并且还需要进行验证。一般来说,研究人员通常使用不同的软件以不同的方式处理数据。整个过程的标准化和通用性对于消除学科障碍和避免交互使用不同软件时的错误至关重要。这需要研究人员通过统一的评估方法,为不同的需求和任务设计标准和合理的管道。
3. 分子指纹和相关步骤选择的任意性。对同一任务使用不同的指纹通常会导致不同的结果。由于缺乏分子指纹性能的黄金标准,很难选择最合适的标准。另外,一些参数,例如通常的相似度阈值的选择,没有明确描述。此外,在选择预测模型时,应保持准确性和可解释性之间的平衡。然而,在大多数研究中,分子指纹、模型和评价指标的选择通常是任意的。
4. 不完整的3D指纹算法。3D分子形状对于生物活性任务至关重要,因为它包含关于分子化学构象的更复杂和全面的信息。然而,在许多情况下,基于2D指纹的方法通常比基于3D形状的方法具有更好的性能。Venkatraman等人比较了五个基于2D的指纹和五个基于3D的指纹的性能,并表明可以从3D指纹中提取的构象特征的数量和构象的灵活性是提高3D方法性能的关键。此外,分子形状和其他化学性质可以结合起来,以开发更复杂和信息丰富的3D模型。
参考文献
Yang J, Cai Y, Zhao K, et al. Concepts and applications of chemical fingerprint for hit and lead screening[J]. Drug Discovery Today, 2022: 103356.
--------- End ---------
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-01-30,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号