🤫 Seurat | 强烈建议收藏的单细胞分析标准流程(经典归一化与降维方法)(二)

🤫 Seurat | 强烈建议收藏的单细胞分析标准流程(经典归一化与降维方法)(二)

生信漫卷
发布于 2023-02-24 13:43:57
发布于 2023-02-24 13:43:57
1写在前面
为了做进一步的分析,我们需要对数据进行归一化(Normalization)和标准化(Z-score)。😘
这里我们介绍一下经典的Normalization方法,这个方法假设所有细胞均含有10,000个UMI。🐶
2用到的包
rm(list = ls())
library(Seurat)
library(tidyverse)
library(SingleR)
library(celldex)
library(RColorBrewer)
library(SingleCellExperiment)
3示例数据
这里我们还是使用之前建好的srat文件,我之前保存成了.Rdata,这里就直接加载了。✌️
load("./srat.Rdata")
srat
4归一化
Note! 这里做的是log-normalization, 大家不要重复取log。🥰
srat <- NormalizeData(srat)

5高变基因
5.1 计算高变基因
找一下高变基因吧,这里补充一下可用的方法:👇
vstmvpdisp
srat <- FindVariableFeatures(srat, selection.method = "vst", nfeatures = 2000)
5.2 Top 10
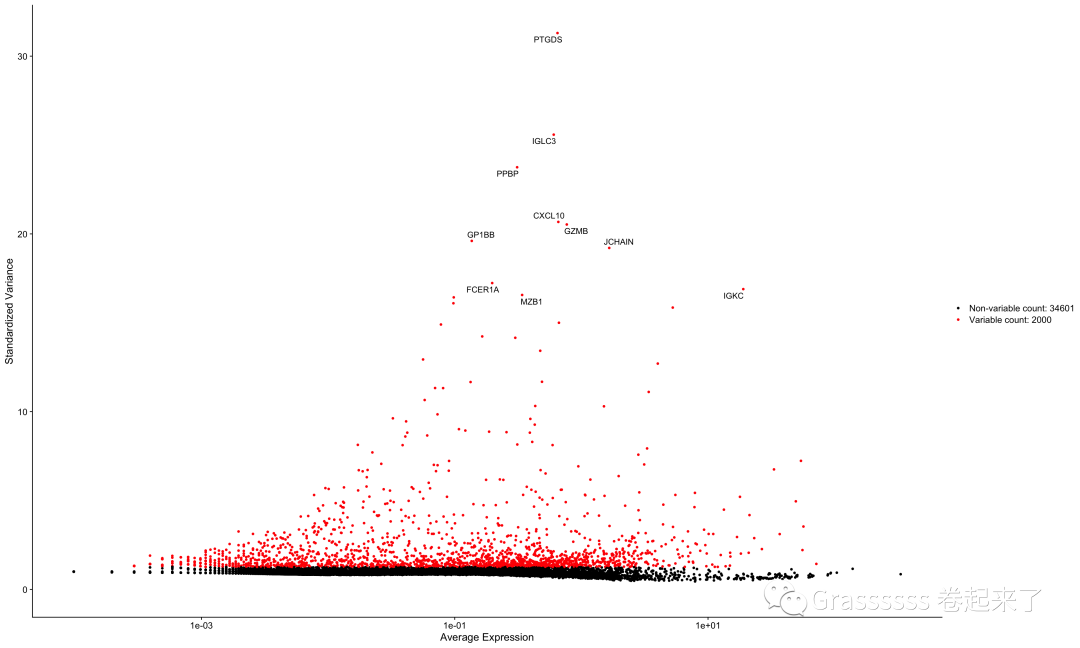
我们取一下高变基因的top10,在后面的可视化中标注出来。
top10 <- head(VariableFeatures(srat), 10)
top10

5.3 可视化
plot1 <- VariableFeaturePlot(srat)
LabelPoints(plot = plot1,
points = top10,
repel = TRUE,
xnudge = 0,
ynudge = 0)
6标准化
细心的小伙伴肯定发现了gene的表达差异较大,这样做出来的图就会不好看。😤
我们在这里接着做一个Z-score, 将Normalization后的数据转为均值为0,标准差为1的数据。🧐
all.genes <- rownames(srat)
srat <- ScaleData(srat, features = all.genes)

7主成分分析
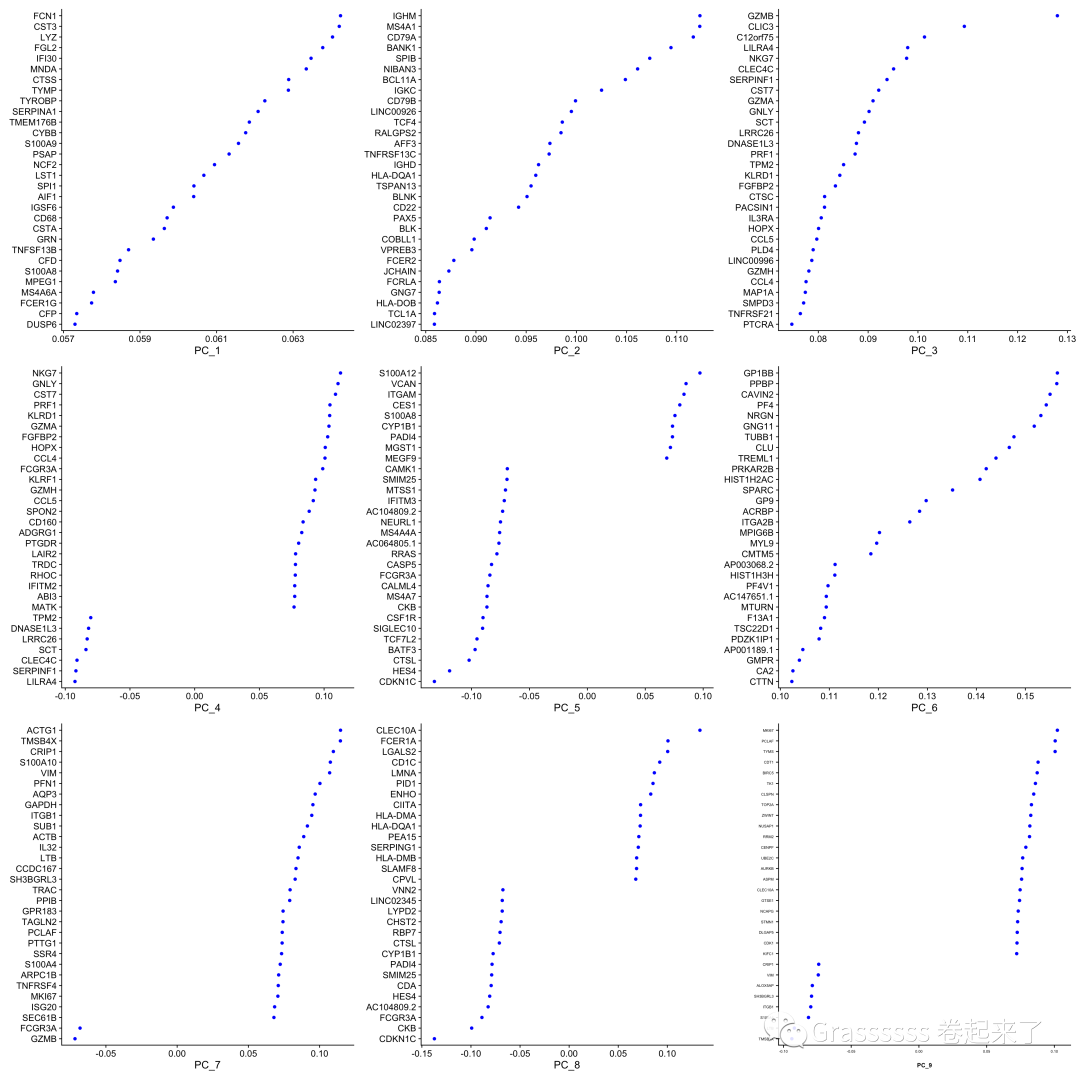
大家可以通过features参数设置纳入PCA的基因数。🌟
这里我们取前2000个高变基因进行分析。
srat <- RunPCA(srat,
features = VariableFeatures(object = srat))
## 可视化
VizDimLoadings(srat, dims = 1:9,
reduction = "pca") +
theme(axis.text=element_text(size=5),
axis.title=element_text(size=8,face="bold"))
当然你也可以用热图的形式来展示结果。😁
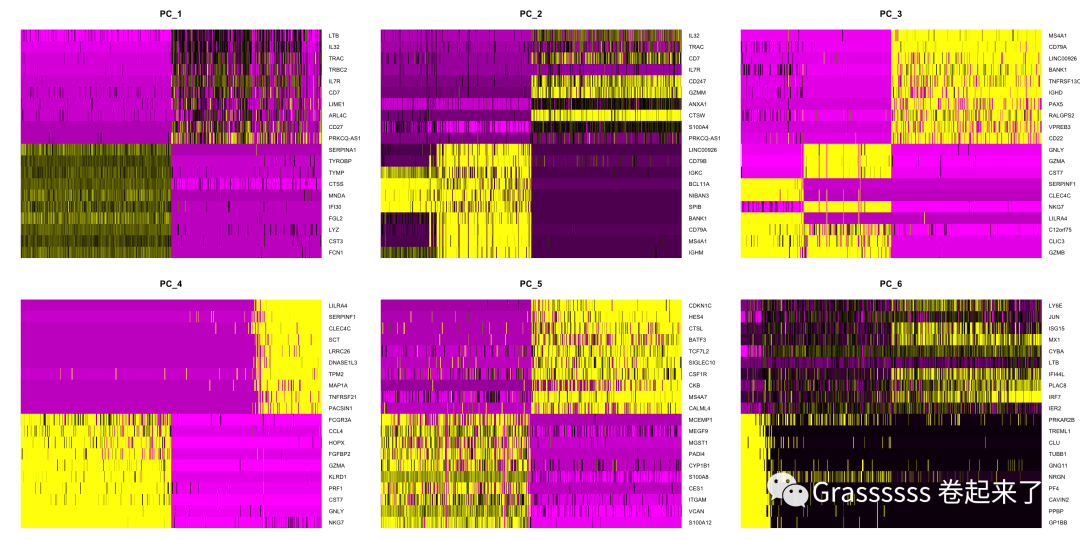
DimHeatmap(srat, dims = 1:6, nfeatures = 20, cells = 500, balanced = T)
8选取多少PC才合适?
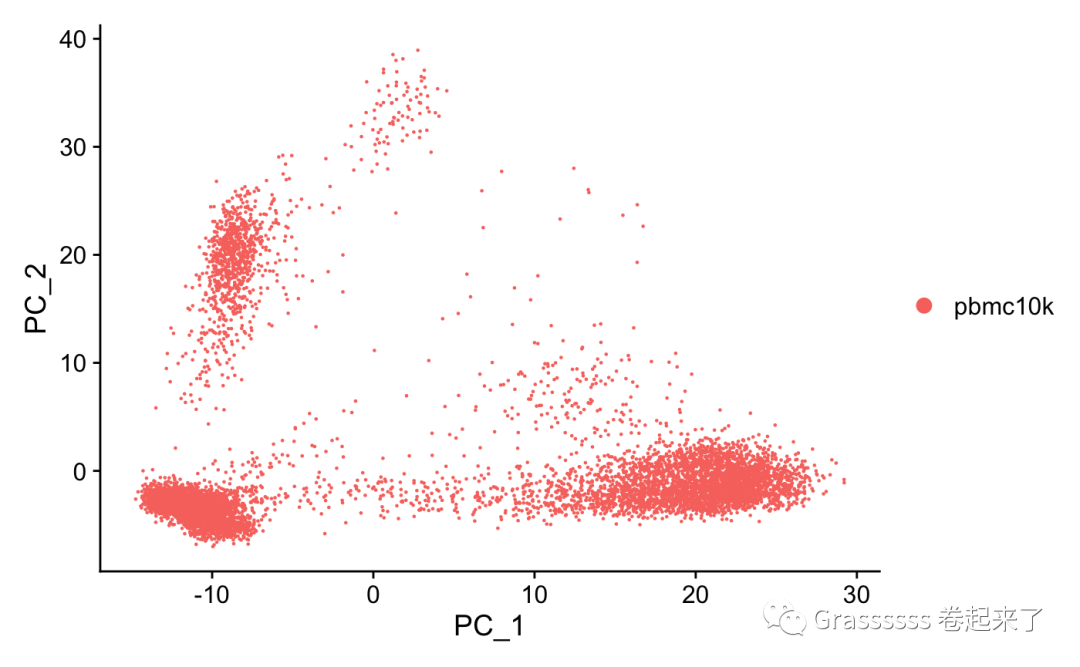
我们先看一下UMAP图。👀
DimPlot(srat) # umap, then tsne, then pca
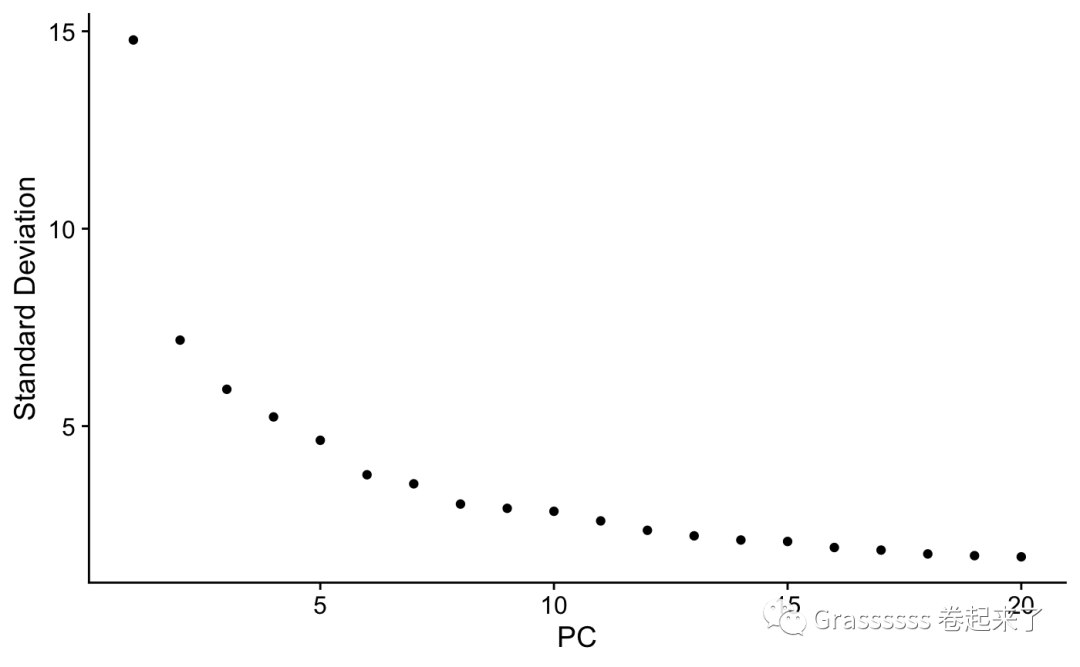
通过ElbowPlot我们可以发现,在10的地方有明显的下降。🤟
ElbowPlot(srat)
9聚类
9.1 开始聚类
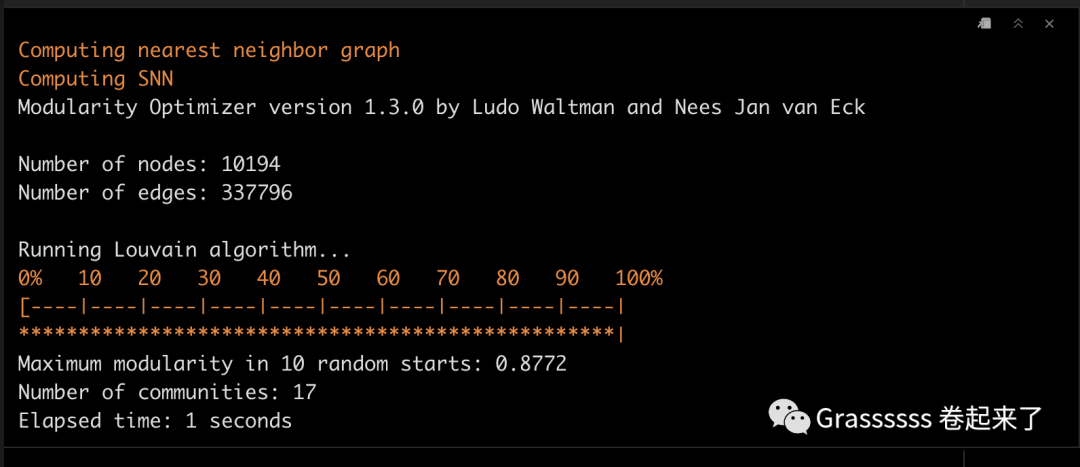
之前确定了10个PCs最合适,这里我们将PC1:10纳入,进行聚类。
这一步非常重要,因为后面marker的选择要基于聚类的结果。 💪
srat <- FindNeighbors(srat, dims = 1:10)
srat <- FindClusters(srat, resolution = 0.8)
UMAP降维。🐹
srat <- RunUMAP(srat, dims = 1:10, verbose = T)
这个时候我们就有一个新的clusters信息了,看一下吧。🙊
table(srat@meta.data$seurat_clusters)

9.2 聚类结果可视化
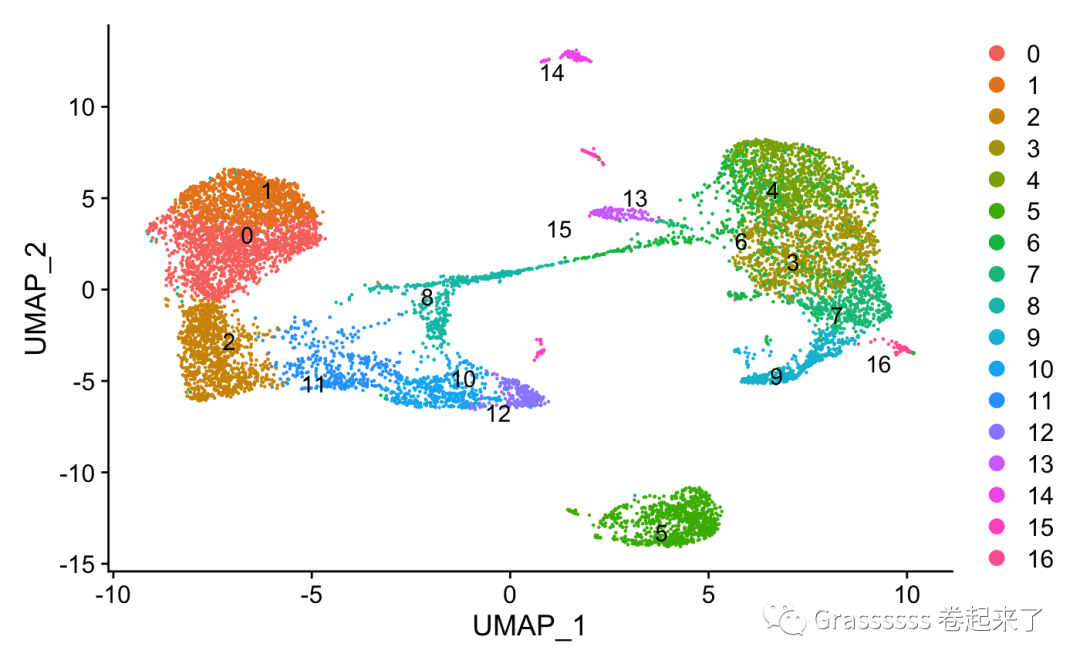
1️⃣ 质控前~
DimPlot(srat,label.size = 4,repel = T,label = T)
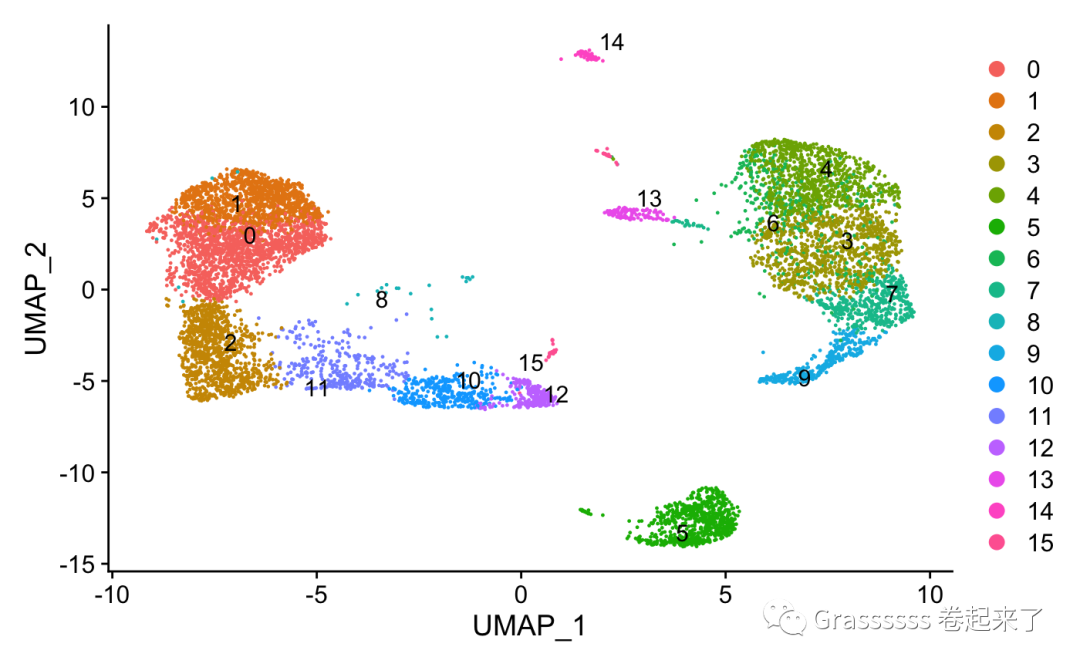
2️⃣ 质控后~
srat <- subset(srat, subset = QC == 'Pass')
DimPlot(srat,label.size = 4,repel = T,label = T)
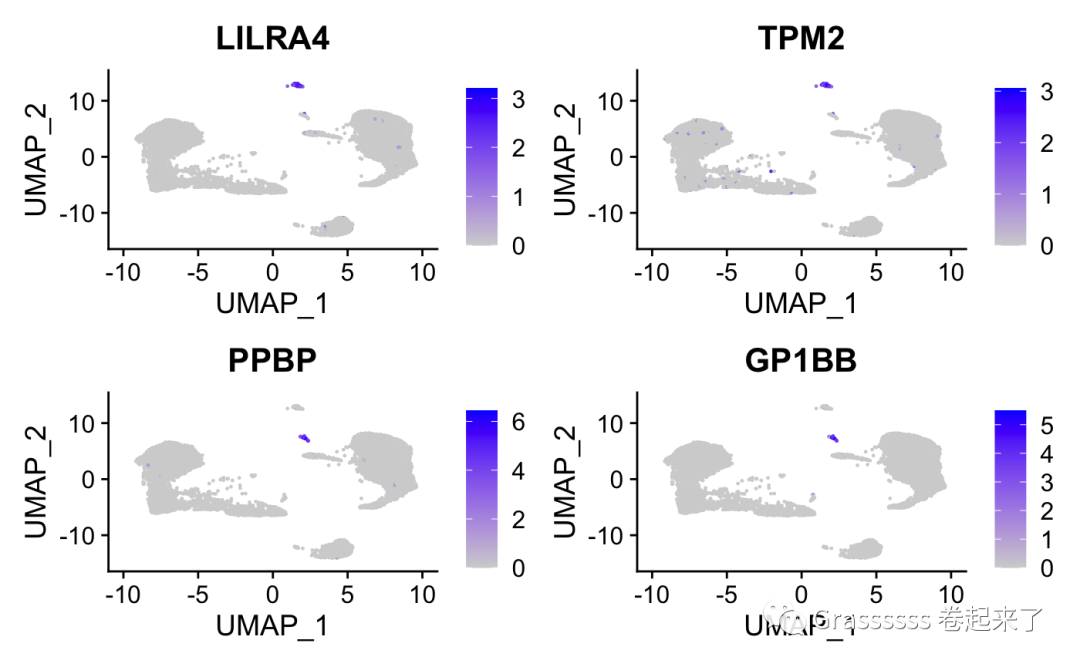
9.3 聚类效果简单评价
这里给大家提供一个简单的评价方法,假设我们已知: 👇
树突状细胞(DCs)的marker是:LILRA4和TPM2;血小板(platelets)的marker是:PPBP和GP1BB。
我们看下这些marker是否都显著表达在一类细胞群中。🤨
FeaturePlot(srat, features = c("LILRA4", "TPM2", "PPBP", "GP1BB"))
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-10-19,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号