免疫浸润结果分子分型(一致性聚类ConsensusClusterPlus)

免疫浸润结果分子分型(一致性聚类ConsensusClusterPlus)

医学和生信笔记
发布于 2023-08-30 13:01:12
发布于 2023-08-30 13:01:12
分子分型也是生信灌水的常见知识点之一。可以用于分子分型的方法非常多,比如:一致性聚类、非负矩阵分解、PCA等等,当然这些方法不需要我们手动去计算,都是有成熟的R包帮我们做。
比较常见的做分子分型的R包:ConsensusClusterPlus, cola, CancerSubtypes, MOVICS, diceR,等等。
可以用于分子分型的数据那更是五花八门啦,理论上只要你有一个数值型矩阵,都可以做分型。
今天给大家演示一个根据免疫浸润结果进行分子分型的示例。
加载数据
我们就用之前批次矫正的数据做演示:批次效应去除之combat和removebatcheffect
rm(list = ls())
load(file = "step1_output.rdata")
expr_combat[1:4,1:4]
## TCGA-D5-6540-01A-11R-1723-07 TCGA-AA-3525-11A-01R-A32Z-07
## MT-CO2 14.50611 14.11911
## MT-CO3 14.43620 13.98267
## MT-ND4 14.36872 13.31916
## MT-CO1 14.61993 13.99882
## TCGA-AA-3525-01A-02R-0826-07 TCGA-AA-3815-01A-01R-1022-07
## MT-CO2 14.04289 14.85734
## MT-CO3 14.20102 14.67482
## MT-ND4 13.51694 14.52215
## MT-CO1 13.62324 14.51841
expr_lnc_combat[1:4,1:4]
## TCGA-D5-6540-01A-11R-1723-07 TCGA-AA-3525-11A-01R-A32Z-07
## MALAT1 5.841875 4.441612
## NORAD 7.840943 6.649570
## SNHG6 7.012464 5.548106
## SNHG29 6.309729 6.020166
## TCGA-AA-3525-01A-02R-0826-07 TCGA-AA-3815-01A-01R-1022-07
## MALAT1 5.579839 6.041946
## NORAD 6.780140 6.638278
## SNHG6 6.435600 6.290823
## SNHG29 8.097017 7.249777
只要肿瘤样本,然后再对表达矩阵做z-score转换,不做也可以,大家可以探索下做与不做的差别。
expr <- expr_combat[,clin_info$sample_type == "tumor"]
expr_lnc <- expr_lnc_combat[,clin_info$sample_type == "tumor"]
clin_info <- clin_info[clin_info$sample_type == "tumor",]
expr[1:4,1:4]
## TCGA-D5-6540-01A-11R-1723-07 TCGA-AA-3525-01A-02R-0826-07
## MT-CO2 14.50611 14.04289
## MT-CO3 14.43620 14.20102
## MT-ND4 14.36872 13.51694
## MT-CO1 14.61993 13.62324
## TCGA-AA-3815-01A-01R-1022-07 TCGA-D5-6923-01A-11R-A32Z-07
## MT-CO2 14.85734 14.47264
## MT-CO3 14.67482 14.13317
## MT-ND4 14.52215 13.75709
## MT-CO1 14.51841 13.93638
expr <- scale(expr)
#save(expr_lnc,file = "step1_expr_lnc.rdata")
免疫浸润分析
还是用之前介绍过的非常好用的IOBR进行ssGSEA分析。
suppressMessages(library(IOBR))
load(file = "G:/bioinfo/000files/ssGSEA28.Rdata")
1行代码即可:
im_ssgsea <- calculate_sig_score(eset = expr
, signature = cellMarker
, method = "ssgsea"
)
##
## >>> Calculating signature score using ssGSEA method
## >>> log2 transformation is not necessary
## Estimating ssGSEA scores for 28 gene sets.
## [1] "Calculating ranks..."
## [1] "Calculating absolute values from ranks..."
##
|
| | 0%
|
|======================================================================| 100%
##
## [1] "Normalizing..."
im_ssgsea[1:4,1:4]
## # A tibble: 4 × 4
## ID `Activated B cell` `Activated CD4 T cell` Activ…¹
## <chr> <dbl> <dbl> <dbl>
## 1 TCGA-3L-AA1B-01A-11R-A37K-07 -0.116 0.0874 0.264
## 2 TCGA-4N-A93T-01A-11R-A37K-07 -0.169 0.0784 0.222
## 3 TCGA-4T-AA8H-01A-11R-A41B-07 -0.273 0.130 0.185
## 4 TCGA-5M-AAT4-01A-11R-A41B-07 -0.326 0.162 0.205
## # … with abbreviated variable name ¹`Activated CD8 T cell`
#save(im_ssgsea,file = "step_ssgsea.rdata")
有了这个结果就可以进行分子分型的操作了。
我们今天介绍的是ConsensusClusterPlus一致性聚类进行分子分型。
一致性聚类
library(ConsensusClusterPlus)
调整下数据格式:
df <- as.data.frame(im_ssgsea)
rownames(df) <- df$ID
df <- df[,-1]
df <- t(df)
df[1:4,1:4]
## TCGA-3L-AA1B-01A-11R-A37K-07
## Activated B cell -0.11564923
## Activated CD4 T cell 0.08738077
## Activated CD8 T cell 0.26405669
## Activated dendritic cell 0.12538222
## TCGA-4N-A93T-01A-11R-A37K-07
## Activated B cell -0.16893409
## Activated CD4 T cell 0.07835561
## Activated CD8 T cell 0.22205116
## Activated dendritic cell 0.10961118
## TCGA-4T-AA8H-01A-11R-A41B-07
## Activated B cell -0.27278589
## Activated CD4 T cell 0.12999000
## Activated CD8 T cell 0.18464372
## Activated dendritic cell 0.09445642
## TCGA-5M-AAT4-01A-11R-A41B-07
## Activated B cell -0.3264560
## Activated CD4 T cell 0.1621332
## Activated CD8 T cell 0.2045764
## Activated dendritic cell 0.1490167
boxplot(df[,1:20])
df1 <- sweep(df,1,apply(df,1,median))#已经做了z-score了,还需要这个中位数归一化的操作吗?大家自己探索下
boxplot(df1[,1:20])

进行一致性聚类,其实就是1行代码:
ccres <- ConsensusClusterPlus(df1,
maxK=9,
reps=100,
pItem=0.8,
pFeature=1,
tmyPal = c("white","#C75D30"),
title='ConsensusCluster/',
clusterAlg="km",
distance="euclidean",
seed=123456,
plot="pdf"
)
## end fraction
## clustered
## clustered
## clustered
## clustered
## clustered
## clustered
## clustered
## clustered
iclres <- calcICL(ccres,title="ics of ssgsea res")
结果会保存到指定目录下的一个PDF文件中,里面有多张图形:
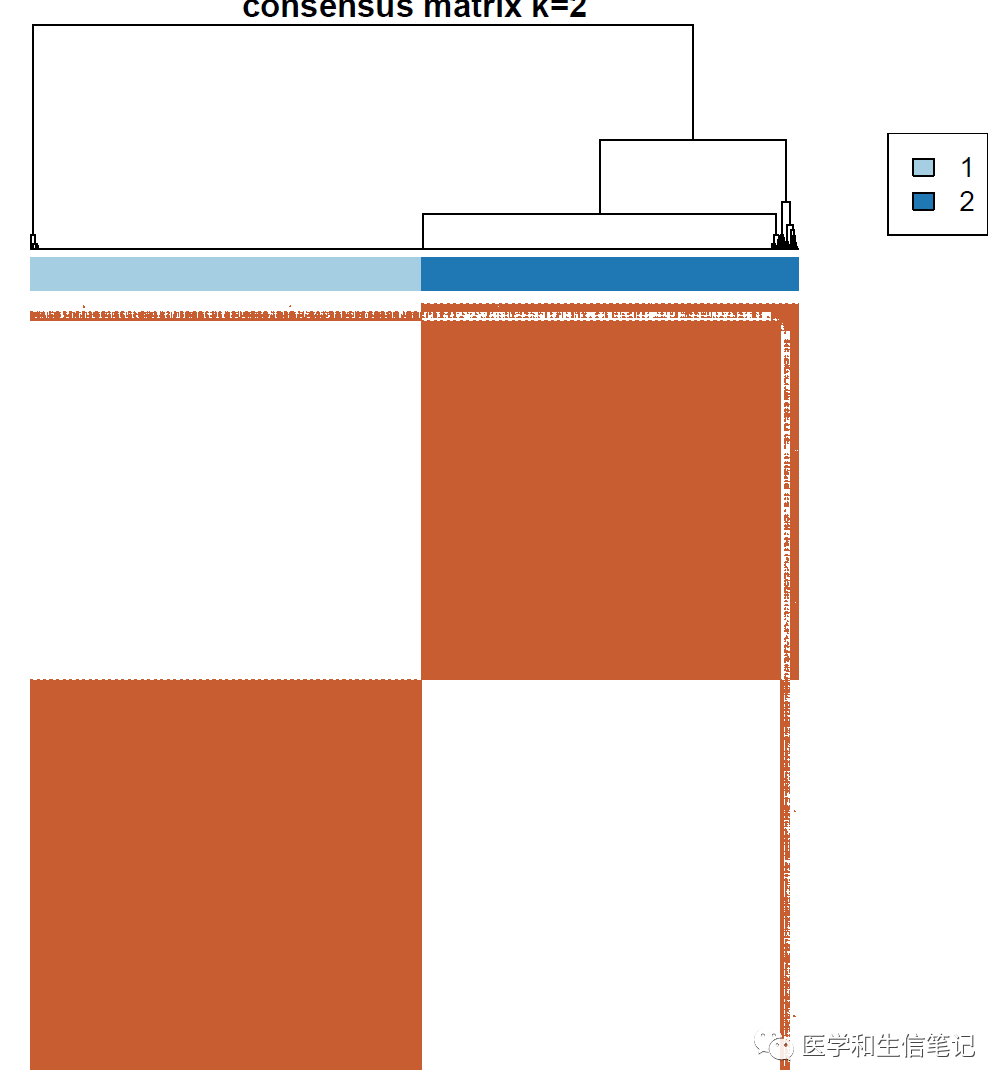
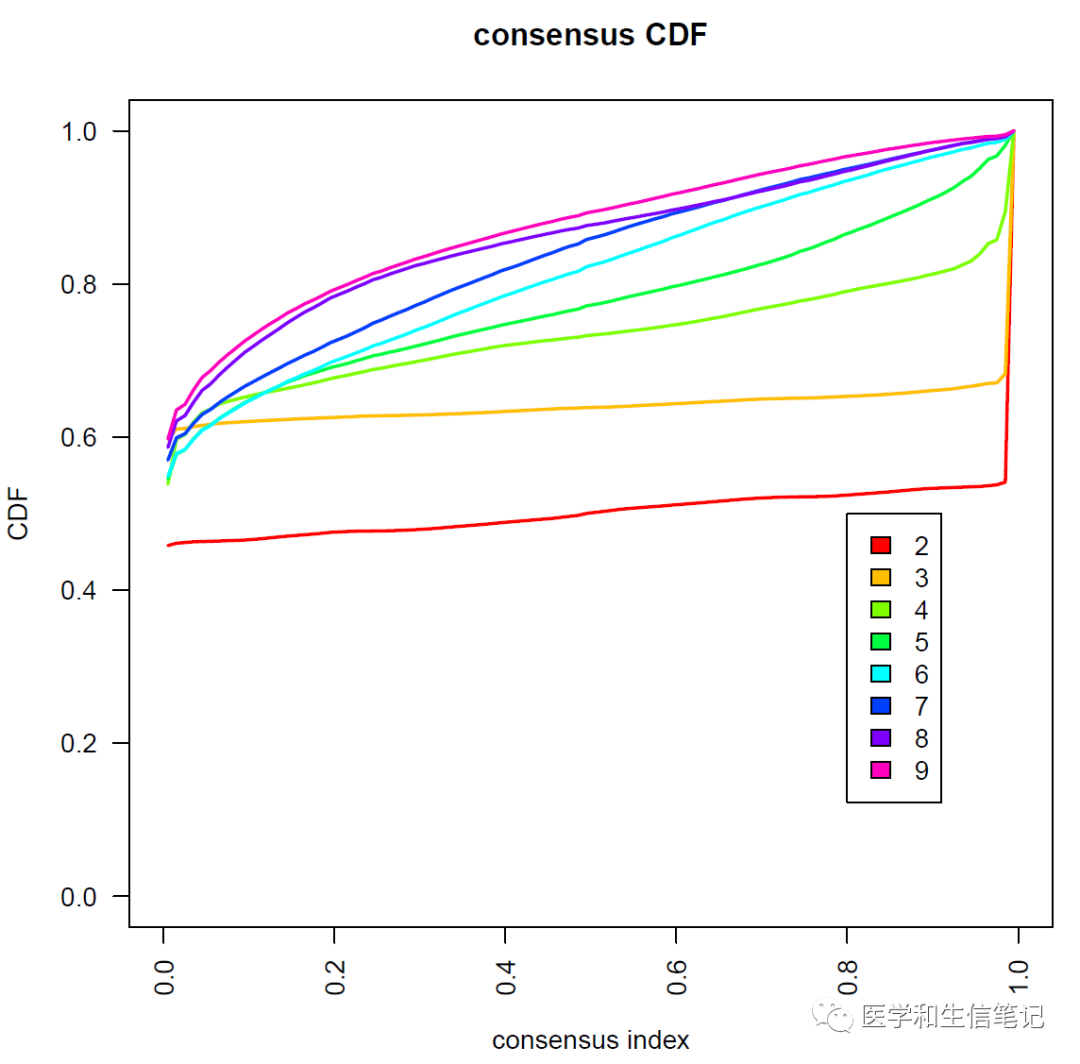
确定最佳聚类个数
标准非常多,比如根据聚类热图看色块干净,没有掺杂;CDF图上升平缓,突然陡峭;delta area拐点(类似于聚类分析的碎石图)
也有大佬根据PAC(模糊聚类比例)确定最佳聚类个数:现在可以直接使用diceR或者cola实现。
## PAC = Proportion of ambiguous clustering 模糊聚类比例
Kvec = 2:9
x1 = 0.1; x2 = 0.9
PAC = rep(NA,length(Kvec))
names(PAC) = paste("K=",Kvec,sep="")
for(i in Kvec){
M = ccres[[i]]$consensusMatrix
Fn = ecdf(M[lower.tri(M)])
PAC[i-1] = Fn(x2) - Fn(x1)
}
optK = Kvec[which.min(PAC)]
optK
## [1] 3
根据PAC和上面一致性聚类给出的图来看,分成3个亚型是最合适的,但是为了演示我们选2!
分型后的数据
根据分型结果提取数据,我们选2:
#提取结果
sample_subtypes <- ccres[[2]][["consensusClass"]]
table(sample_subtypes)
## sample_subtypes
## 1 2
## 331 319
331个样本是第1型,319个样本是第2型。
免疫浸润箱线图
这个数据的样本顺序和ssGSEA结果的样本顺序是完全一致的,可以直接用,所以我们就根据这个分型,探索下不同亚型的免疫浸润情况:
suppressMessages(library(tidyverse))
im_ssgsea %>%
mutate(sample_subtypes = factor(sample_subtypes)) %>%
pivot_longer(-c(ID,sample_subtypes), names_to = "cell_type",values_to = "value") %>%
ggplot(aes(cell_type,value,fill=sample_subtypes))+
geom_boxplot()+
labs(x=NULL)+
theme(legend.position = "top",axis.text.x = element_text(angle = 45,hjust = 1))

这种箱线图大家可能已经审美疲劳了,我给大家介绍一种更好看的小提琴图,当然我是从文献里看到的,但是代码来自于stackoverflow。使用关键词搜索即可得到这段代码。
首先我们自己定义一个GeomSplitViolin,然后就可以和ggplot2对接使用了!
# https://stackoverflow.com/questions/47651868/split-violin-plot-with-ggplot2-with-quantiles
library(ggplot2)
GeomSplitViolin <- ggproto("GeomSplitViolin", GeomViolin,
draw_group = function(self, data, ..., draw_quantiles = NULL) {
# Original function by Jan Gleixner (@jan-glx)
# Adjustments by Wouter van der Bijl (@Axeman)
data <- transform(data, xminv = x - violinwidth * (x - xmin), xmaxv = x + violinwidth * (xmax - x))
grp <- data[1, "group"]
newdata <- plyr::arrange(transform(data, x = if (grp %% 2 == 1) xminv else xmaxv), if (grp %% 2 == 1) y else -y)
newdata <- rbind(newdata[1, ], newdata, newdata[nrow(newdata), ], newdata[1, ])
newdata[c(1, nrow(newdata) - 1, nrow(newdata)), "x"] <- round(newdata[1, "x"])
if (length(draw_quantiles) > 0 & !scales::zero_range(range(data$y))) {
stopifnot(all(draw_quantiles >= 0), all(draw_quantiles <= 1))
quantiles <- create_quantile_segment_frame(data, draw_quantiles, split = TRUE, grp = grp)
aesthetics <- data[rep(1, nrow(quantiles)), setdiff(names(data), c("x", "y")), drop = FALSE]
aesthetics$alpha <- rep(1, nrow(quantiles))
both <- cbind(quantiles, aesthetics)
quantile_grob <- GeomPath$draw_panel(both, ...)
ggplot2:::ggname("geom_split_violin", grid::grobTree(GeomPolygon$draw_panel(newdata, ...), quantile_grob))
}
else {
ggplot2:::ggname("geom_split_violin", GeomPolygon$draw_panel(newdata, ...))
}
}
)
create_quantile_segment_frame <- function(data, draw_quantiles, split = FALSE, grp = NULL) {
dens <- cumsum(data$density) / sum(data$density)
ecdf <- stats::approxfun(dens, data$y)
ys <- ecdf(draw_quantiles)
violin.xminvs <- (stats::approxfun(data$y, data$xminv))(ys)
violin.xmaxvs <- (stats::approxfun(data$y, data$xmaxv))(ys)
violin.xs <- (stats::approxfun(data$y, data$x))(ys)
if (grp %% 2 == 0) {
data.frame(
x = ggplot2:::interleave(violin.xs, violin.xmaxvs),
y = rep(ys, each = 2), group = rep(ys, each = 2)
)
} else {
data.frame(
x = ggplot2:::interleave(violin.xminvs, violin.xs),
y = rep(ys, each = 2), group = rep(ys, each = 2)
)
}
}
geom_split_violin <- function(mapping = NULL, data = NULL, stat = "ydensity", position = "identity", ...,
draw_quantiles = NULL, trim = TRUE, scale = "area", na.rm = FALSE,
show.legend = NA, inherit.aes = TRUE) {
layer(data = data, mapping = mapping, stat = stat, geom = GeomSplitViolin, position = position,
show.legend = show.legend, inherit.aes = inherit.aes,
params = list(trim = trim, scale = scale, draw_quantiles = draw_quantiles, na.rm = na.rm, ...))
}
接下来就画图即可,是不是还挺别致的:
im_ssgsea %>%
mutate(sample_subtypes = factor(sample_subtypes)) %>%
pivot_longer(-c(ID,sample_subtypes), names_to = "cell_type",values_to = "value") %>%
ggplot(aes(cell_type,value,fill=sample_subtypes))+
geom_split_violin(draw_quantiles = c(0.25, 0.5, 0.75))+
theme_bw()+
theme(legend.position = "top",
axis.text.x = element_text(angle = 45,hjust = 1)
)
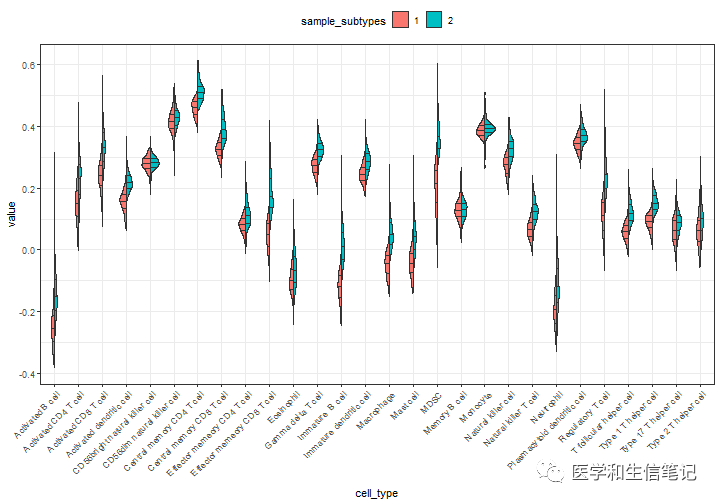
#保存下数据
save(clin_info,sample_subtypes,im_ssgsea,file = "step2_output.rdata")
样本信息热图
有了这个分子亚型的数据,再结合其他临床数据,我们就可以画出一个热图,综合展现不同类型样本的免疫浸润情况。
我们选择生存状态,年龄,性别,病理分期,MSI这几个临床信息进行展示。
clin_sub <- clin_info[,c("vital_status","age_at_diagnosis","gender","ajcc_pathologic_stage","paper_MSI_status")]
clin_sub$sample_cluster <- sample_subtypes
names(clin_sub) <- c("status","age","gender","stage","msi","cluster")
clin_sub$msi <- as.character(clin_sub$msi)
str(clin_sub)
## 'data.frame': 650 obs. of 6 variables:
## $ status : chr "Alive" "Alive" "Alive" "Alive" ...
## $ age : int 24282 32871 23922 21118 23825 26541 15842 22574 29194 30237 ...
## $ gender : chr "male" "male" "female" "male" ...
## $ stage : chr "Stage I" "Stage IIIB" "Stage IIA" "Stage I" ...
## $ msi : chr NA "MSI-H" "MSI-H" NA ...
## $ cluster: int 1 1 1 1 1 2 1 1 2 2 ...
table(clin_sub$stage)
##
## Stage I Stage IA Stage II Stage IIA Stage IIB Stage IIC Stage III
## 110 1 38 185 14 2 25
## Stage IIIA Stage IIIB Stage IIIC Stage IV Stage IVA Stage IVB
## 15 85 60 65 24 2
table(clin_sub$msi)
##
## MSI-H MSI-L MSS Not Evaluable
## 41 46 196 1
对这几个信息重新整理一下:
# 把NA变成字符型的“NA”,方便后面使用!
clin_sub[is.na(clin_sub)] <- 'NA'
# 亚型重新编码为c1 c2
# 年龄变为 >65 <=65 "NA">23725 TRUE
# 病理分期变为1,2,3,4期
# 然后都变成因子型
clin_sub <- clin_sub %>%
mutate(cluster = ifelse(cluster == 1, "c1","c2"),
msi = ifelse(msi=="Not Evaluable","NA",msi),
age = ifelse(age == "NA","NA",ifelse(age>23725,">65","<=65")),
stage = case_when(stage %in% c("Stage I","Stage IA") ~ "I",
stage %in% c("Stage II","Stage IIA","Stage IIB","Stage IIC") ~ "II",
stage %in% c("Stage III","Stage IIIA","Stage IIIB","Stage IIIC") ~ "III",
stage %in% c("Stage IV","Stage IVA","Stage IVB") ~ "IV",
.default = stage
)
) %>%
mutate(age = factor(age,levels = c('<=65','>65',"NA")),
stage = factor(stage, levels=c('I','II','III','IV',"NA")),
gender = factor(gender, levels=c('female','male',"NA")),
status = factor(status, levels=c("Alive","Dead", "NA" )),
msi=factor(msi, levels=c("MSI-H","MSI-L","MSS","NA"))
)
glimpse(clin_sub)
## Rows: 650
## Columns: 6
## $ status <fct> Alive, Alive, Alive, Alive, Alive, Alive, Alive, Dead, Alive, …
## $ age <fct> >65, >65, >65, <=65, >65, >65, <=65, <=65, >65, >65, >65, >65,…
## $ gender <fct> male, male, female, male, male, male, male, female, female, fe…
## $ stage <fct> I, III, II, I, III, I, III, III, III, III, III, IV, III, III, …
## $ msi <fct> NA, MSI-H, MSI-H, NA, NA, MSI-L, NA, NA, NA, MSI-H, NA, NA, MS…
## $ cluster <chr> "c1", "c1", "c1", "c1", "c1", "c2", "c1", "c1", "c2", "c2", "c…
identical(rownames(clin_sub),im_ssgsea$ID)
## [1] FALSE
让免疫浸润结果的样本顺序和我们准备的临床信息的样本顺序一致,方便使用:
clin_sub <- clin_sub[match(im_ssgsea$ID, rownames(clin_sub)),]
identical(rownames(clin_sub),im_ssgsea$ID)
## [1] TRUE
把样本这一列变成行名:
ssgsea_df <- column_to_rownames(im_ssgsea,"ID")
然后就可以画图,本来准备使用tidyHeatmap的,但是注释条中不能有缺失值,差评!
还是要使用画热图最强大的R包:ComplexHeatmap,靠谱!
# 不能有NA
#library(tidyHeatmap)
library(ComplexHeatmap)
columnAnno <- HeatmapAnnotation(status = clin_sub$status,
age = clin_sub$age,
gender = clin_sub$gender,
stage = clin_sub$stage,
msi = clin_sub$msi,
cluster = clin_sub$cluster
#,na_col = "white"
)
scaled_ssgsea <- scale(t(ssgsea_df))
scaled_ssgsea[scaled_ssgsea>2] <- 2
scaled_ssgsea[scaled_ssgsea< -2] <- -2
ComplexHeatmap::Heatmap(scaled_ssgsea, na_col = "white",show_column_names = F,
row_names_side = "left",name = "fraction",
column_order = c(rownames(ssgsea_df)[c(grep("c1",clin_sub$cluster),grep("c2",clin_sub$cluster))]),
column_split = clin_sub$cluster, column_title = NULL,
cluster_columns = F,
top_annotation = columnAnno
)

这个信息还是很全面的,这种热图也是各类生信文章中常见的图形。
estimate评估免疫纯度
我们还可以使用其他方法评价一下不同亚型的免疫浸润情况,每种方法都试一下,增加可信度和工作量......
这里我们就选择estimate
# 还是使用estimate,1行代码即可
estires <- deconvo_estimate(expr, platform = "illumina")
##
## >>> Running ESTIMATE
## [1] "Merged dataset includes 9883 genes (529 mismatched)."
## [1] "1 gene set: StromalSignature overlap= 136"
## [1] "2 gene set: ImmuneSignature overlap= 140"
# 调整样本顺序
estires <- estires[match(rownames(clin_sub),estires$ID),]
identical(estires$ID,rownames(clin_sub))
## [1] TRUE
# 加上亚型信息
estires$cluster <- clin_sub$cluster
# 简单画个图
ggplot(estires, aes(cluster, ImmuneScore_estimate))+
geom_boxplot()+
ggpubr::stat_compare_means(aes(group = cluster,label = ..p.signif..),
method = "wilcox.test")+
theme_classic()

结果并不显著哦~大家可以尝试其他免疫浸润方法,都看看,这样就可以把显著的结果放在文章里了!
save(expr, clin_info, clin_sub, im_ssgsea, file = "step3_output.rdata")
有了这个分型后,你还可以根据这个分型做各种分析,比如生存分析、差异分析、富集分析等等,反正就是查看两种亚型之间的各种差别以及和各种临床信息的联系,我就不再演示了,大家自己尝试下即可。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-07-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号