【孟德尔随机化】工具变量对表型的解释率R^2


R^2,表型解释率(phenotypic variance explained, PVE),
method 1
- Computing proportion of variance in phenotype explained by a given SNP (PVE) [1]

要注意哦,这里的(se(β))^2其实是standard error的平方,不关β的事儿。
这个公式应该是最为大家所熟知的,但是有个问题,根据这个公式计算所得的R^2,是针对单个SNP的。如果我的表型相关工具变量不止一个呢,所有工具变量对表型的解释率又该如何计算呢?有的文章是把单个SNP的R^2相加[2],但我没找到这个方法的依据,暂且按下不表。
PS:researchgate论坛上面大家对于这个公式也有不同的看法。
How to determine the percent phenotypic variation explained (PVE) by a selected SNP? | ResearchGate How to calculate proportion of phenotypic variance explained by GWAS significant SNP? | ResearchGate
也有人在文章中运用了这个公式[3],一起来看看——

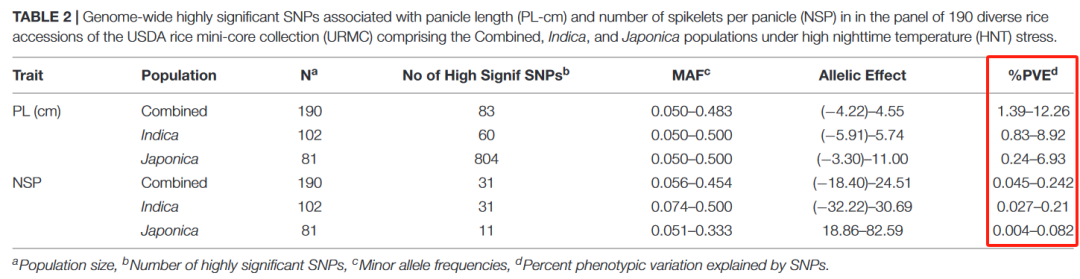
得到的PVE值还是针对单个SNP的PVE
method 2
【GWAS】如何计算显著关联位点的表型解释率PVE(phenotypic variation explained)?- 生物信息与育种 - 博客园 (cnblogs.com)
熟悉的TwoSampleMR包

但是这个方法计算得到的R^2和第一种方法得到的值不太一样~
Exposure$R2 <- (2 * (Exposure$BETA^2) * Exposure$AF1 * (1 - Exposure$AF1)) /
(2 * (Exposure$BETA^2) * Exposure$AF1 * (1 - Exposure$AF1) +
2 * Exposure$N * Exposure$AF1 * (1 - Exposure$AF1)*Exposure$SE^2)
Exposure$NR2 <- get_r_from_bsen(Exposure$BETA,Exposure$SE,Exposure$N)
目前没有看到用这个方法计算R^2的文章,如果大家看到的话,欢迎在评论区讨论呀~
method 3
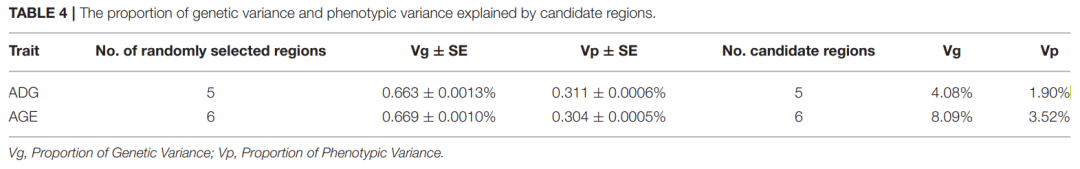
候选区域所解释的遗传变异和表型变异的比例是通过多随机效应混合线性模型估算的。The model can be written as[4]——

method 4
文章原文:这些分析确定了 26 个位点的次要信号;当这些额外的 SNP 与主要 SNP 相结合时,映射变异的总集合可以解释弗莱明汉姆心脏研究中每种血脂性状总变异的 12.4%(TC)、12.2%(LDL-C)、12.1%(HDL-C)和 9.6%(TG),相当于每种性状遗传变异的 25%~30%[5]。
计算公式在Supplementary Materials
method 5
[6]

method 6
文献是这么说明自己的结果的:The significant SNPs in our study explained up to 58 % of the variance .[7]
是总的SNPs对于表型的解释率✌,看看怎么计算得到的⬇
SNP contribution to the phenotypic variance was estimated using ANOVA with the R package:

┭┮﹏┭┮,太难了,除了方法一和二,其他的方法都对我都不太友好。
method 7
How to calculate the amount of phenotype variance explained by a few SNPs? | ResearchGate
在这个帖子里,有一位大佬给了非常详细(但我还是看不太懂)的方案,如果大家有兴趣可以再研究一下~
目前关于R^2的计算方法还是比较多的,大家可以自由挑选以上的方法进行计算。以上也仅仅介绍了目前了解到的几种,如果对于这方面的知识有更深入的学习需求,推荐大家再看这两篇文献⬇
- Pitfalls of predicting complex traits from SNPs
- A review of SNP heritability estimation methods
参考文献
[1] Shim H, Chasman DI, Smith JD, Mora S, Ridker PM, Nickerson DA, Krauss RM, Stephens M. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One. 2015 Apr 21;10(4):e0120758. doi: 10.1371/journal.pone.0120758. PMID: 25898129; PMCID: PMC4405269.
[2] Deng Y, Ge W, Xu H, Zhang J. A Mendelian randomization study of the effect of tea intake on breast cancer. Front Nutr. 2022;9:956969. Published 2022 Oct 18. doi:10.3389/fnut.2022.956969
[3] Kumar A, Gupta C, Thomas J, Pereira A. Genetic Dissection of Grain Yield Component Traits Under High Nighttime Temperature Stress in a Rice Diversity Panel. Front Plant Sci. 2021;12:712167. Published 2021 Sep 28. doi:10.3389/fpls.2021.712167
[4] Tang Z, Xu J, Yin L, et al. Genome-Wide Association Study Reveals Candidate Genes for Growth Relevant Traits in Pigs. Front Genet. 2019;10:302. Published 2019 Apr 5. doi:10.3389/fgene.2019.00302
[5] Teslovich TM, Musunuru K, Smith AV, , et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010 Aug 5;466(7307):707-13. doi: 10.1038/nature09270. PMID: 20686565; PMCID: PMC3039276.
[6] Nagelkerke, N. J. D. (1991). A Note on a General Definition of the Coefficient of Determination. Biometrika, 78(3), 691–692. https://doi.org/10.2307/2337038
[7] Zhao, K., Tung, CW., Eizenga, G. et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2, 467 (2011). https://doi.org/10.1038/ncomms1467
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-10-10,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号