单细胞转录组学轨迹分析解析3-Slingshot代码解析
原创
单细胞转录组学轨迹分析解析3-Slingshot代码解析
原创
小胡子刺猬的生信学习123
发布于 2023-12-05 17:22:11
发布于 2023-12-05 17:22:11
前面一节对单细胞轨迹的数据过滤和降维已经做了解析,而其实主要用这个软件的是用后面的拟时序分析的内容。因此下面对拟时序的内容进行解析。
3 Using Slingshot
基于此,我们已拥有在模拟数据集上运行Slingshot所需的准备文件。主要分为两步,包括使用基于聚类的最小生成树 (MST) 识别全局谱系结构,并拟合同步主曲线来描述每个谱系。
这两个步骤可以与 getLineages 和 getCurves 函数一起运行,也可以与包装函数 slingshot 一起运行。我们将使用包装函数来分析单轨迹数据集,但稍后在分叉数据集上演示各个函数的用法。
Slingshot包装函数在单个调用中执行轨迹推理的两个步骤。必要的输入是坐标的降维矩阵和一组聚类标签。这些对象可以是单独的对象,或者在单轨迹数据的情况下,是包含在 SingleCellExperiment 对象中的元素。
为了使用 PCA 产生的降维和高斯混合建模识别的聚类标签来运行Slingshot,我们将进行以下操作:
sce <- slingshot(sce, clusterLabels = 'GMM', reducedDim = 'PCA')如上所述,如果未提供聚类结果,则假定所有单元格都是同一聚类的一部分,并且将构建一条曲线。如果未提供降维,则 slingshot 将使用 reducedDims 返回的列表中的第一个元素。
输出是一个 SingleCellExperiment 对象,其中包含Slingshot结果。所有结果都存储在 PseudotimeOrdering 对象中,该对象被添加到原始对象的 colData 中,可以通过 colData(sce)$slingshot 查询。此外,所有推断的伪时间变量(每个谱系一个)都会单独添加到 colData 中。要将所有Slingshot结果提取到单个对象中,我们可以使用 as。PseudotimeOrdering 或 as.SlingshotDataSet 函数,具体取决于我们想要它的形式。PseudotimeOrdering 对象是 SummarizedExperiment 对象的扩展,这些对象是灵活的容器,可用于大多数用途。SlingshotDataSet 对象主要用于可视化,因为包中包含多种绘图方法。下面,我们将单轨迹数据的推断谱系可视化,并用伪时间着色的点。
summary(sce$slingPseudotime_1)## Min. 1st Qu. Median Mean 3rd Qu. Max. ## 0.000 8.631 21.121 21.414 34.363 43.185library(grDevices)colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)plotcol <- colors[cut(sce$slingPseudotime_1, breaks=100)] plot(reducedDims(sce)$PCA, col = plotcol, pch=16, asp = 1)lines(SlingshotDataSet(sce), lwd=2, col='black')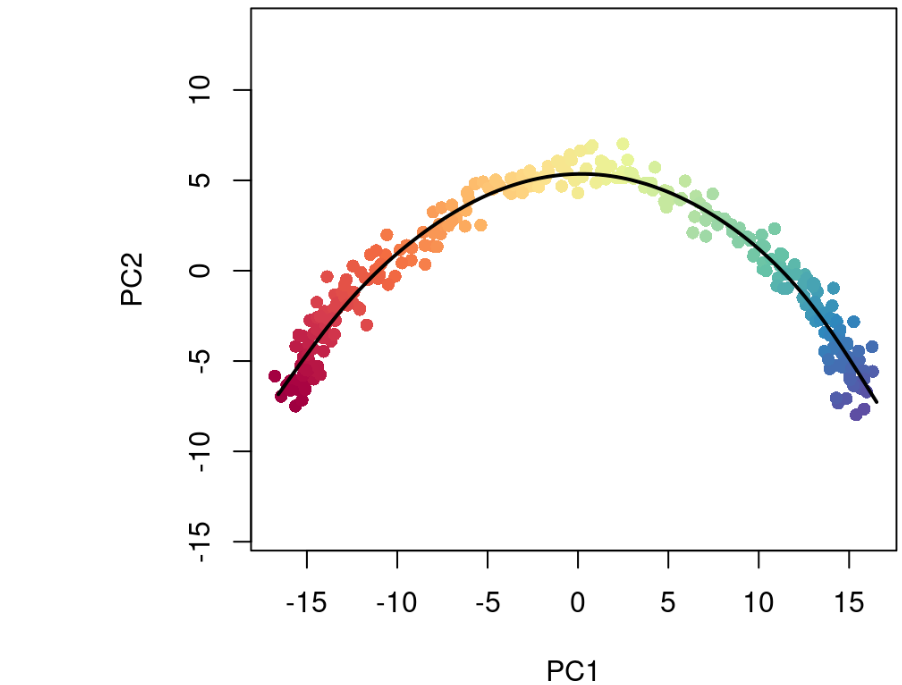
我们还可以看到基于聚类的最小生成树如何使用类型参数直观地估计世系结构。
plot(reducedDims(sce)$PCA, col = brewer.pal(9,'Set1')[sce$GMM], pch=16, asp = 1)lines(SlingshotDataSet(sce), lwd=2, type = 'lineages', col = 'black')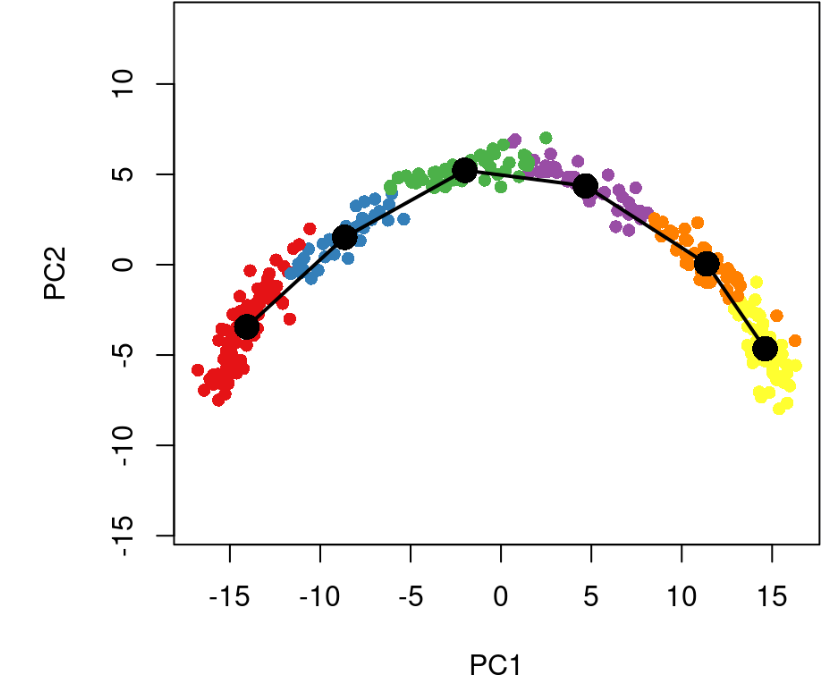
4 Downstream Analysis
4.1 Identifying temporally dynamic genes
在运行弹弓之后,我们通常对寻找在发育过程中改变其表达的基因感兴趣。我们将使用tradeSeq 软件包(Van den Berge et al., 2020)演示这种类型的分析。
对于每个基因,我们将使用负二项式噪声分布拟合一个通用加性模型 (GAM),以模拟基因表达和伪时间之间的(潜在非线性)关系。然后,我们将使用 associationTest 测试表达式和伪时间之间的显著关联。
library(tradeSeq)# fit negative binomial GAMsce <- fitGAM(sce)# test for dynamic expressionATres <- associationTest(sce)然后,我们可以根据 p 值挑选出排名靠前的基因,并使用热图可视化它们在发育过程中的表达。在这里,我们使用前 250 个最动态表达的基因。
topgenes <- rownames(ATres[order(ATres$pvalue), ])[1:250]pst.ord <- order(sce$slingPseudotime_1, na.last = NA)heatdata <- assays(sce)$counts[topgenes, pst.ord]heatclus <- sce$GMM[pst.ord]heatmap(log1p(heatdata), Colv = NA, ColSideColors = brewer.pal(9,"Set1")[heatclus])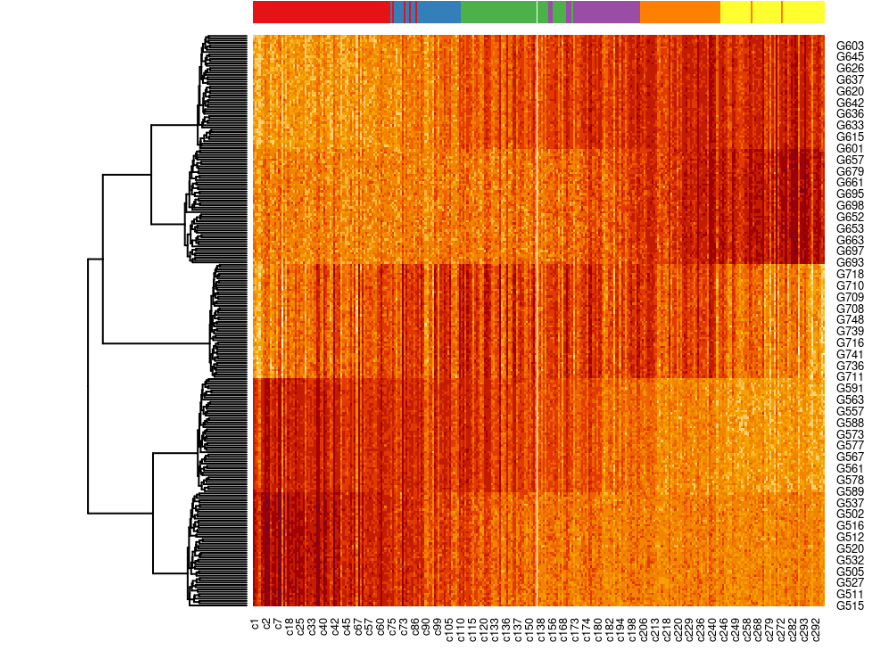
5 Detailed Slingshot Functional
在这里,我们提供了更多详细信息,并重点介绍了Slingshot包的一些附加功能。我们将使用包含的 slingshotExample 数据集进行说明。该数据集旨在表示低维空间中的细胞,并带有一组由 k 生成的聚类标签-表示聚类。我们将直接使用低维坐标矩阵,并提供聚类标签作为附加参数,而不是构建一个需要基因水平数据的完整 SingleCellExperiment 对象。
5.1 Identifying global lineage structure
getLineages 函数将 n 个×作为输入p 矩阵和长度为 n 的聚类结果向量。它使用最小生成树 (MST) 映射相邻群集之间的连接,并标记通过这些连接表示世系的路径。此函数的输出是一个 PseudotimeOrdering,其中包含输入以及推断的 MST(由 igraph 对象表示)和谱系(簇名称的有序向量)。
这种分析可以以完全无监督的方式执行,也可以通过指定已知的初始点和终点聚类以半监督方式执行。如果我们没有指定起点,slingshot 会根据简约原则选择一个起点,从而最大限度地提高拆分前世系之间共享的簇数。如果没有拆分或多个聚类产生相同的精简分数,则任意选择起始聚类。
对于我们的模拟数据,slingshot 选择聚类 1 作为起始聚类。然而,我们通常建议根据先验知识(样本采集时间或已建立的基因标记)来规范初始簇。此规范不会影响 MST 的构造方式,但会影响分支曲线的构造方式。
lin1 <- getLineages(rd, cl, start.clus = '1')lin1## class: PseudotimeOrdering## dim: 140 2## metadata(3): lineages mst slingParams## pathStats(2): pseudotime weights## cellnames(140): cell-1 cell-2 ... cell-139 cell-140## cellData names(2): reducedDim clusterLabels## pathnames(2): Lineage1 Lineage2## pathData names(0):plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)lines(SlingshotDataSet(lin1), lwd = 3, col = 'black')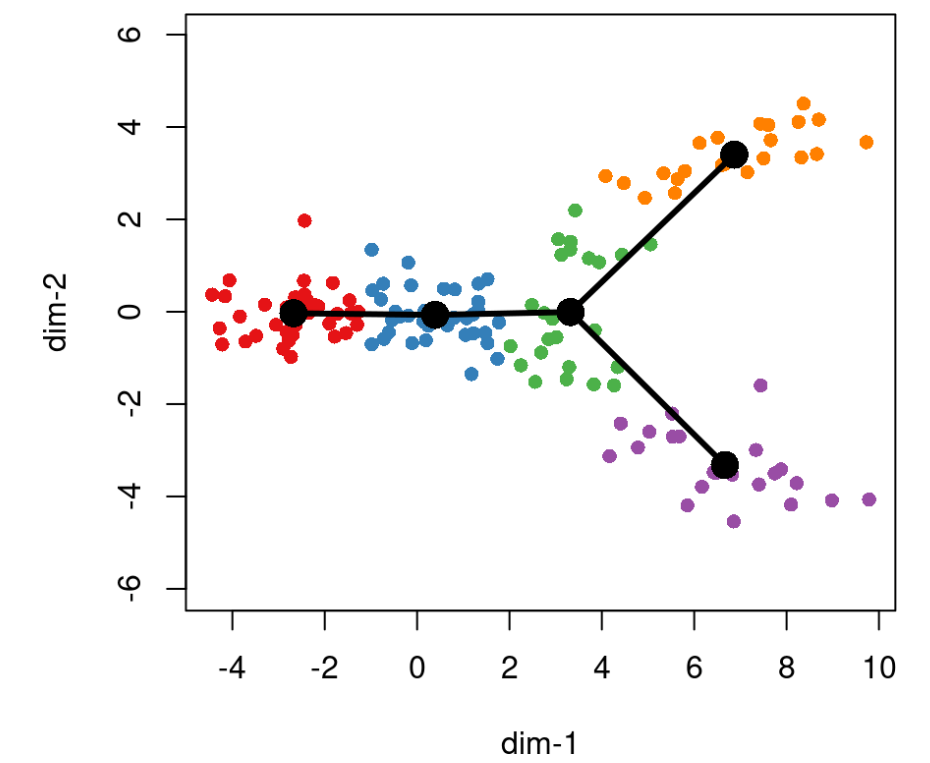
在此步骤中,slingshot 还允许指定已知端点。在构造 MST 时,指定为终端单元状态的群集将被限制为只有一个连接(即,它们必须是叶节点)。此约束可能会影响树的其他部分的绘制方式,如下一个示例所示,其中我们将集群 3 指定为终点。
lin2 <- getLineages(rd, cl, start.clus= '1', end.clus = '3')plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)lines(SlingshotDataSet(lin2), lwd = 3, col = 'black', show.constraints = TRUE)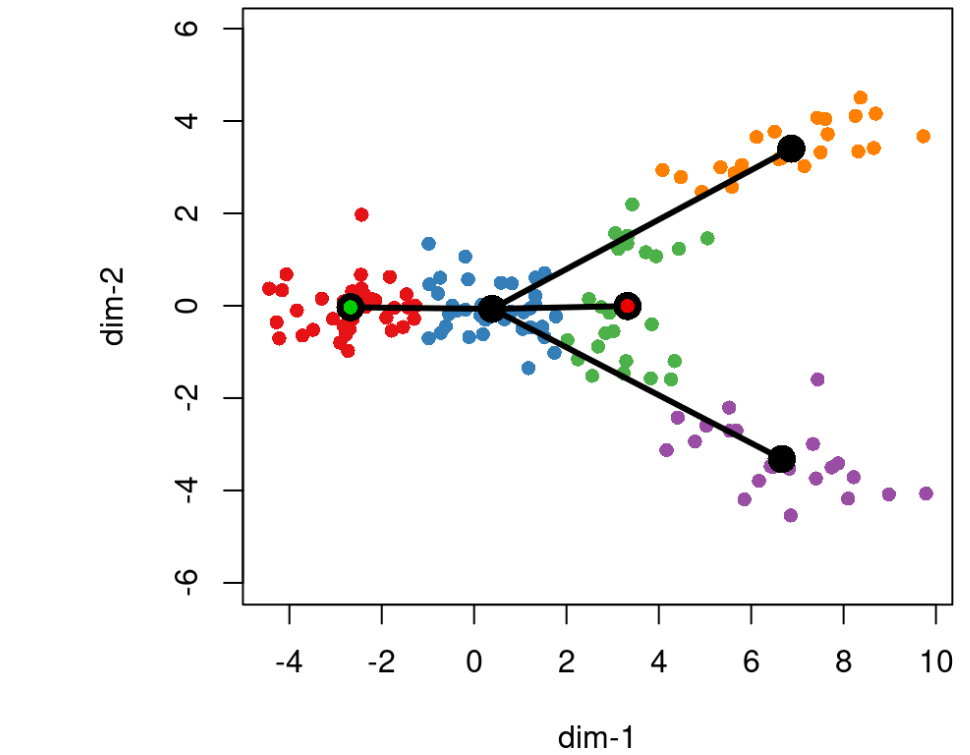
5.2 Constructing smooth curves and ordering cells
为了沿着这些不同的谱系进行建模,我们将使用函数 getCurves 构建平滑曲线。使用基于所有单元的平滑曲线可以消除单元投影到分段线性轨迹顶点上的问题,并使slingshot对聚类结果中的噪声更具robust。
当有两个或多个谱系时,我们会在算法中添加一个额外的步骤:平均共享单元格附近的曲线。两个谱系在尚未分化的细胞上应该相当一致,因此在每次迭代中,我们平均这些细胞附近的曲线。这增加了算法的稳定性并产生平滑的分支谱系。
crv1 <- getCurves(lin1)crv1## class: PseudotimeOrdering## dim: 140 2## metadata(4): lineages mst slingParams curves## pathStats(2): pseudotime weights## cellnames(140): cell-1 cell-2 ... cell-139 cell-140## cellData names(2): reducedDim clusterLabels## pathnames(2): Lineage1 Lineage2## pathData names(0): plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)lines(SlingshotDataSet(crv1), lwd = 3, col = 'black')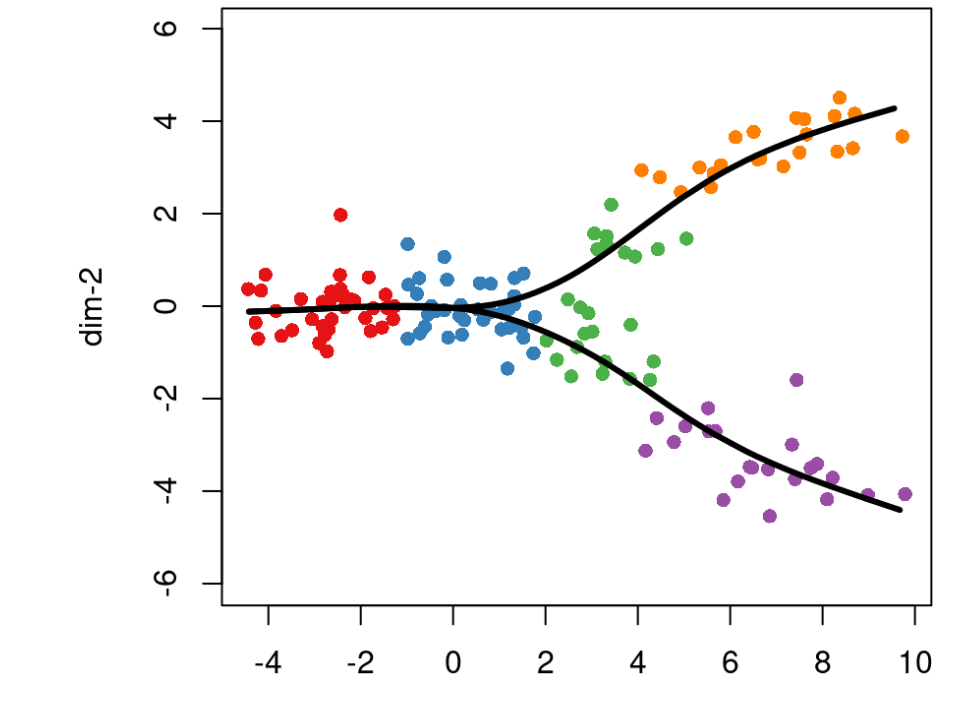
5.3 Running Slingshot on large datasets
对于大型数据集,我们强烈建议将 approx_points 参数与 slingshot(或 getCurves)一起使用。这允许用户指定曲线的分辨率(即唯一点的数量)。虽然 MST 构造在聚类上运行,但随着数据集大小的增长,将所有点迭代投影到一条或多条曲线上的过程可能会变得计算繁重。因此,我们将 approx_points 的默认值设置为 150或数据集中的单元格数,以较小者为准。这将大大降低探索性分析的计算成本,同时对结果轨迹的影响最小
sce5 <- slingshot(sce, clusterLabels = 'GMM', reducedDim = 'PCA', approx_points = 5)colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)plotcol <- colors[cut(sce5$slingPseudotime_1, breaks=100)]plot(reducedDims(sce5)$PCA, col = plotcol, pch=16, asp = 1)lines(SlingshotDataSet(sce5), lwd=2, col='black')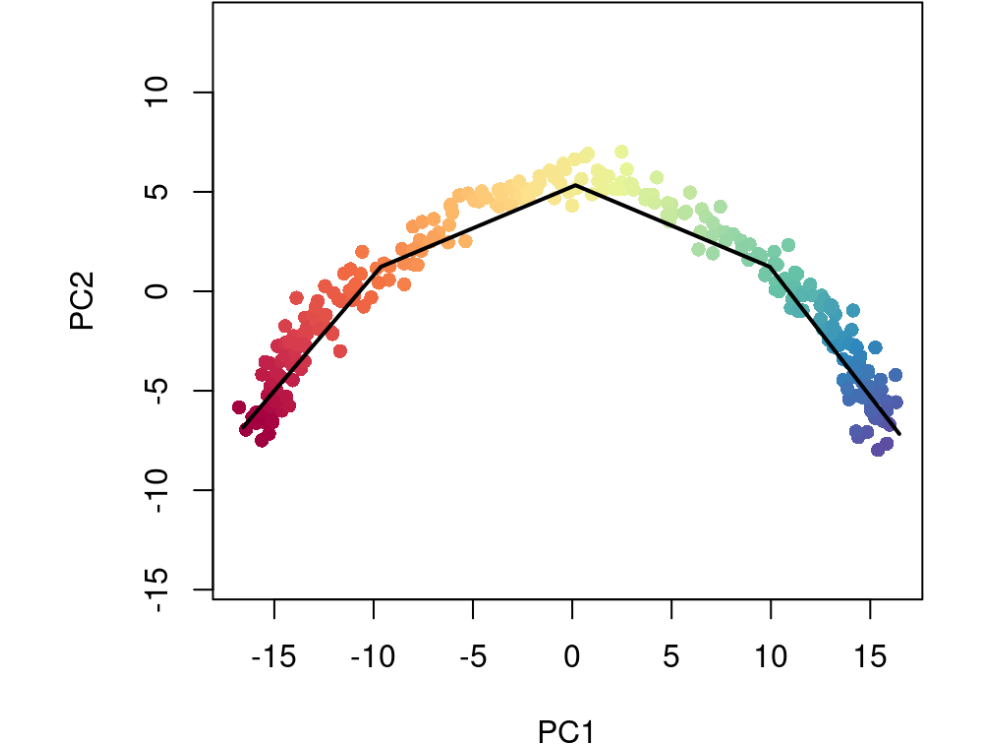
5.4 Multiple Trajectories
在某些情况下,我们感兴趣的是识别多个不相交的轨迹。Slingshot在最初的MST构造中通过引入一个称为omega的人工集群来解决这个问题。这个人为簇与每个实簇之间有固定的长度,这意味着任何两个实簇之间的最大距离是这个长度的两倍。实际上,这为 MST 中允许的最大边长设置了限制。设置 omega = TRUE 将实现经验法则,即最大允许边长等于 3乘以在没有人工聚类的情况下构造的 MST 的中值边长(注意:这相当于说 omega_scale 的默认值为 1.5。
rd2 <- rbind(rd, cbind(rd[,2]-12, rd[,1]-6))cl2 <- c(cl, cl + 10)pto2 <- slingshot(rd2, cl2, omega = TRUE, start.clus = c(1,11))plot(rd2, pch=16, asp = 1, col = c(brewer.pal(9,"Set1"), brewer.pal(8,"Set2"))[cl2])lines(SlingshotDataSet(pto2), type = 'l', lwd=2, col='black')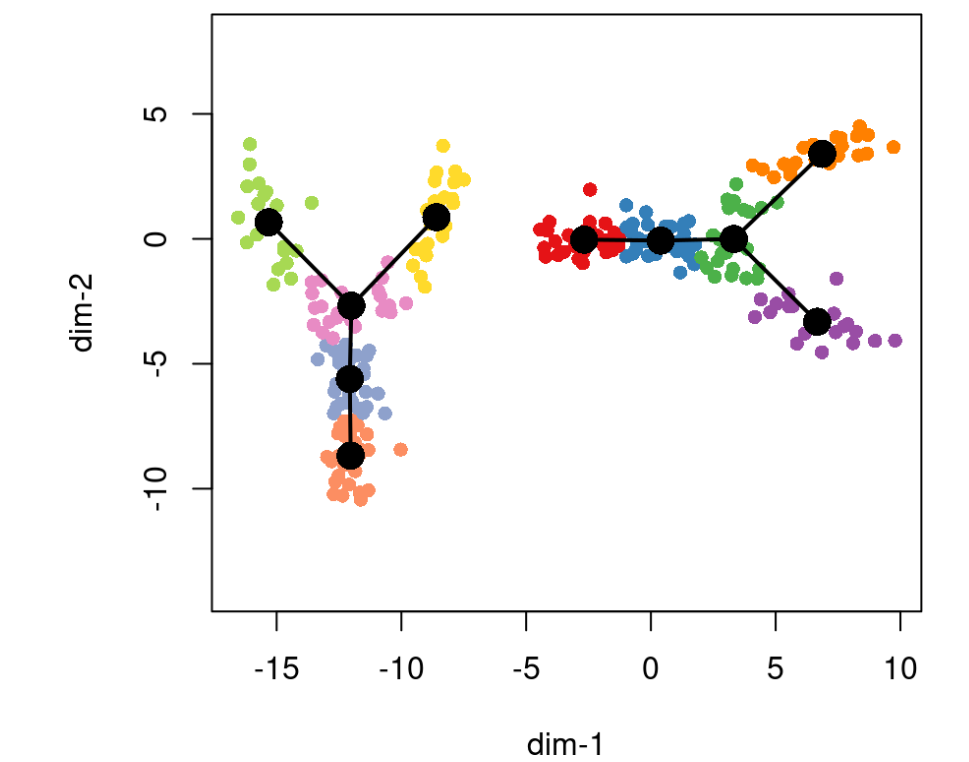
随后Slingshot也可以进行多个曲线分别可视化。
plot(rd2, pch=16, asp = 1, col = c(brewer.pal(9,"Set1"), brewer.pal(8,"Set2"))[cl2])lines(SlingshotDataSet(pto2), lwd=2, col='black')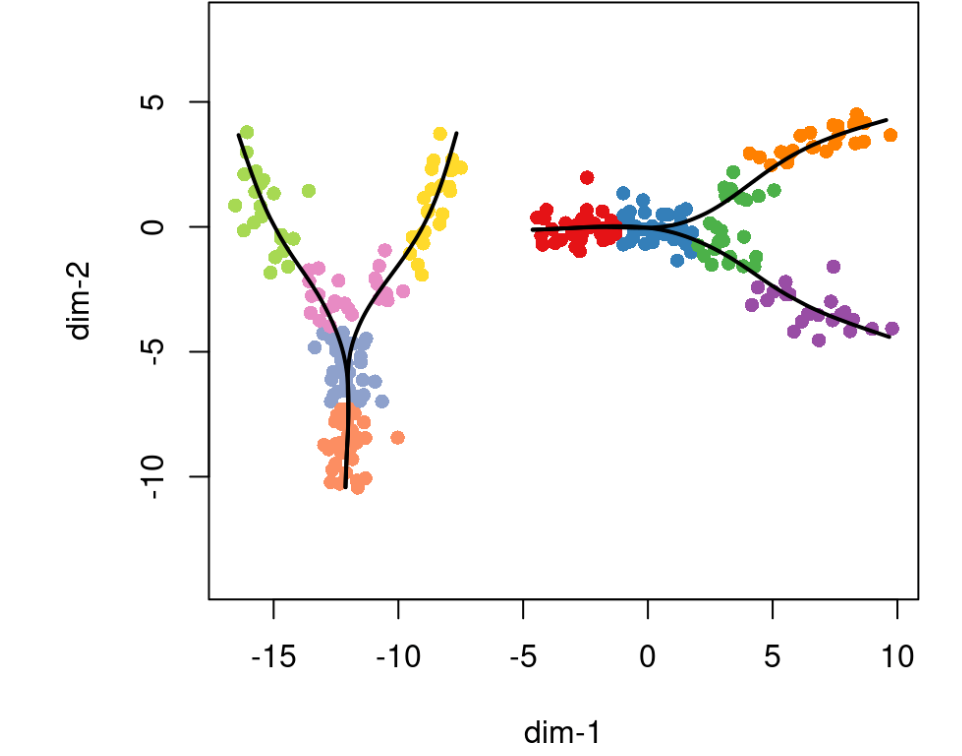
5.5 Projecting Cells onto Existing Trajectories
有时,我们可能只想使用单元格的子集来确定轨迹,或者我们可能会获得想要投影到现有轨迹上的新数据。无论哪种情况,我们都需要一种方法来确定新细胞沿着先前构建的轨迹的位置。为此,我们可以使用 predict 函数。
# our original PseudotimeOrderingpto <- sce$slingshot # simulate new cells in PCA spacenewPCA <- reducedDim(sce, 'PCA') + rnorm(2*ncol(sce), sd = 2) # project onto trajectorynewPTO <- slingshot::predict(pto, newPCA)newplotcol <- colors[cut(slingPseudotime(newPTO)[,1], breaks=100)]plot(reducedDims(sce)$PCA, col = 'grey', bg = 'grey', pch=21, asp = 1, xlim = range(newPCA[,1]), ylim = range(newPCA[,2]))lines(SlingshotDataSet(sce), lwd=2, col = 'black')points(slingReducedDim(newPTO), col = newplotcol, pch = 16)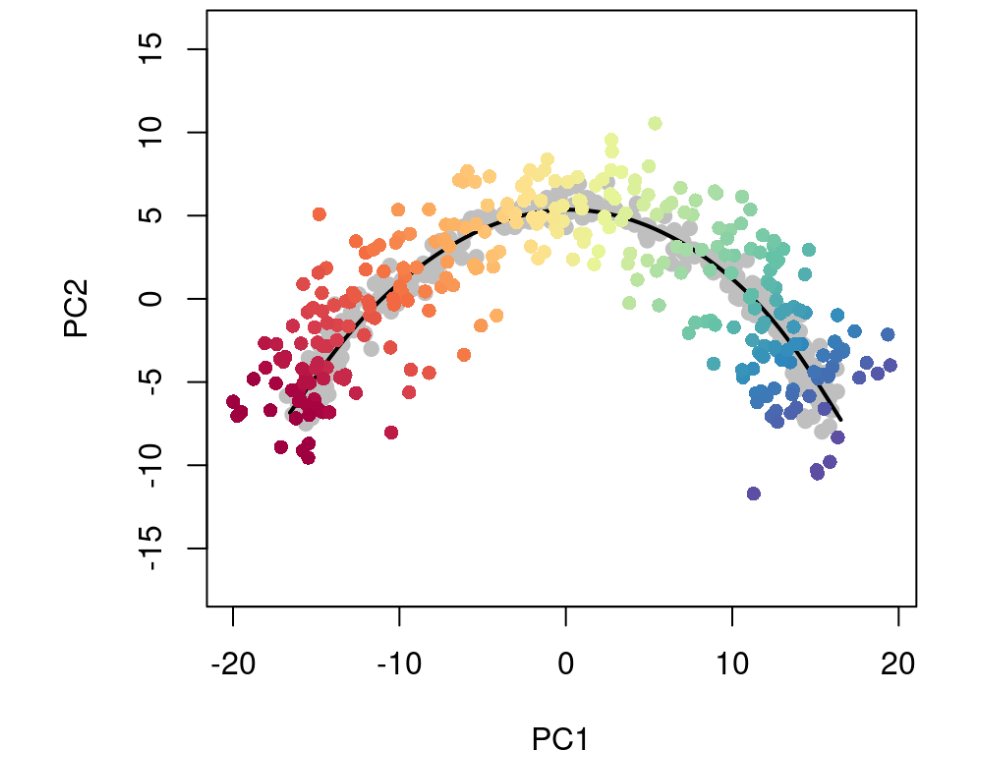
参考文献
Van den Berge, K., Roux de Bézieux, H., Street, K., Saelens, W., Cannoodt, R., Saeys, Y., Dudoit, S., & Clement, L. (2020). Trajectory-based differential expression analysis for single-cell sequencing data. Nature Communications, 11(1), 1201. https://doi.org/10.1038/s41467-020-14766-3
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号