跟着PNAS学数据分析:泛基因组(pan-genome)分析核心基因组可变基因组大小

跟着PNAS学数据分析:泛基因组(pan-genome)分析核心基因组可变基因组大小

用户7010445
发布于 2023-12-19 15:18:56
发布于 2023-12-19 15:18:56
论文
Novel functional sequences uncovered through a bovine multiassembly graph
代码链接
https://github.com/AnimalGenomicsETH/bovine-graphs/tree/main
代码是一个snakemake流程,慢慢看,争取把每个rule都拆解出来
基本思路
使用minigraph做基因组比对,获得一个图基因组,图基因组包含边和节点,节点是序列。然后把每个基因组单独比对回图基因组,可以判断图基因组中节点是否被覆盖,如果所有基因组都覆盖这个节点,这个节点就是核心基因组的一部分,否则就是可变基因组
这里需要理解一下gfa格式的文件
论文提供了分析流程用到的代码,我们用拟南芥的数据试试,拟南芥的论文
Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics
https://www.nature.com/articles/s41467-020-14779-y
这个论文里就做了核心基因组和可变基因组的分析,但是这里的方法和PNAS牛的这篇文章不一样
代码
7个拟南芥基因组序列,只用组装到染色体水平的序列
seqkit grep -r -f chr.list ../../An-1.chr.all.v2.0.fasta -o 00.assembly/An1.fa
seqkit grep -r -f chr.list ../../C24.chr.all.v2.0.fasta -o 00.assembly/C24.fa
seqkit grep -r -f chr.list ../../Cvi.chr.all.v2.0.fasta -o 00.assembly/Cvi.fa
seqkit grep -r -f chr.list ../../Eri.chr.all.v2.0.fasta -o 00.assembly/Eri.fa
seqkit grep -r -f chr.list ../../Kyo.chr.all.v2.0.fasta -o 00.assembly/Kyo.fa
seqkit grep -r -f chr.list ../../Ler.chr.all.v2.0.fasta -o 00.assembly/Ler.fa
seqkit grep -r -f chr.list ../../Sha.chr.all.v2.0.fasta -o 00.assembly/Sha.fa
minigraph构建图基因组,参考基因组放第一个位置
time minigraph --inv no -xggs -t 48 \
00.assembly/An1.fa 00.assembly/C24.fa \
00.assembly/Cvi.fa 00.assembly/Eri.fa \
00.assembly/Kyo.fa \
00.assembly/Ler.fa \
00.assembly/Sha.fa > at.gfa
8m50.144s
minigraph再把每个基因组比对到图基因组上
这里gaf和gfa有啥区别?
minigraph -t 48 --cov -x asm at.gfa 00.assembly/An1.fa > An1.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/C24.fa > C24.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/Cvi.fa > Cvi.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/Eri.fa > Eri.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/Kyo.fa > Kyo.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/Ler.fa > Ler.gaf
minigraph -t 48 --cov -x asm at.gfa 00.assembly/Sha.fa > Sha.gaf
从gaf文件中解析每个node的覆盖信息
python comb_coverage01.py -g An1.gaf -a An1 -o An1Cov.tsv -r Y
python comb_coverage01.py -g C24.gaf -a C24 -o C24Cov.tsv -r N
python comb_coverage01.py -g Cvi.gaf -a Cvi -o CviCov.tsv -r N
python comb_coverage01.py -g Eri.gaf -a Eri -o EriCov.tsv -r N
python comb_coverage01.py -g Ler.gaf -a Ler -o LerCov.tsv -r N
python comb_coverage01.py -g Sha.gaf -a Sha -o ShaCov.tsv -r N
合并数据
library(tidyverse)
read_tsv("D:/Jupyter/PNAS_bovine/An1Cov.tsv") %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/C24Cov.tsv"),
by=c("nodeid"="nodeid")) %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/CviCov.tsv"),
by=c("nodeid"="nodeid")) %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/EriCov.tsv"),
by=c("nodeid"="nodeid")) %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/KyoCov.tsv"),
by=c("nodeid"="nodeid")) %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/LerCov.tsv"),
by=c("nodeid"="nodeid")) %>%
left_join(read_tsv("D:/Jupyter/PNAS_bovine/ShaCov.tsv"),
by=c("nodeid"="nodeid")) %>%
select(-c(2:5)) -> datmat
datmat %>%
column_to_rownames("nodeid") -> datmat
datmat[datmat>0] <- 1
datmat %>%
rownames_to_column("nodeid") %>%
write_tsv("D:/Jupyter/PNAS_bovine/nodemat.tsv")
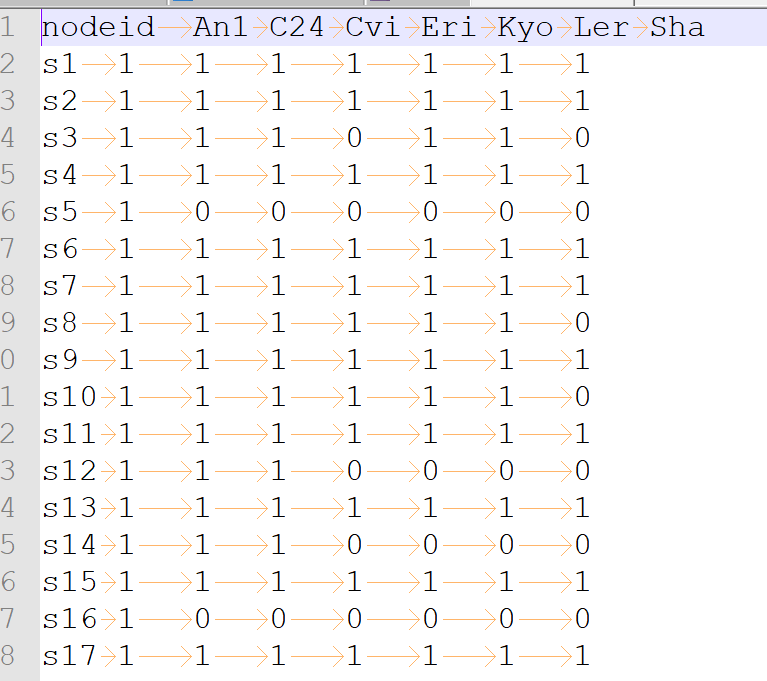
image.png
awk '$1~/S/ {{ split($5,chr,":"); split($6,pos,":"); split($7,arr,":");print $2,length($3),chr[3],pos[3],arr[3] }}' at.gfa > graph_len.tsv
这段awk的代码不是太明白,对比输出和输入能看出来是在做啥
核心基因组大小
nodemat<-"D:/Jupyter/PNAS_bovine/nodemat.tsv"
datmat <- read.table(nodemat, header = TRUE, stringsAsFactors = FALSE)
datmat$combres <- rowSums(datmat %>% select(-nodeid))
datmat
totassemb <- ncol(datmat) - 2
totassemb
graphlen<-"D:/Jupyter/PNAS_bovine/graph_len.tsv"
datlen <- read.table(graphlen, header = FALSE, stringsAsFactors = FALSE)
colnames(datlen) <- c("nodeid", "conlen", "chromo", "pos", "rrank")
datlen
datmat <- datmat %>% left_join(datlen, by = c("nodeid"))
datmat %>% head()
core <- sum(datmat[datmat$combres == totassemb, "conlen"])
core
这个是95M,论文中写的是105M
泛基因组曲线
datexp <- read.table(nodemat, header = TRUE, stringsAsFactors = FALSE)
datexp
datlen <- read.table(graphlen, header = FALSE, stringsAsFactors = FALSE)
datlen %>% head()
colnames(datlen) <- c("nodeid", "conlen", "chromo", "pos", "rrank")
breeds <- colnames(datexp %>% select(-nodeid))
breeds
no_rep <- ncol(datexp) - 1
no_rep
datpan <- data.frame(
norep = numeric(),
nosamp = numeric(),
assemb = character(),
core_gen = numeric(),
flex_gen = numeric(),
tot_gen = numeric()
)
datcon <- datlen %>% select(nodeid, conlen)
datcon
for (nosamp in seq(1, length(breeds))) {
# how many sampling repeated
for (norep in seq(1, no_rep)) {
selsamp <- as.character(sample(breeds, size = nosamp))
# selected breeds
selcol <- datexp[, colnames(datexp) %in% selsamp, drop = FALSE]
# add contig length
# it is ordered so we just add it from conlen
# add number of colour in node
selcol$comcol <- rowSums(selcol)
# add contig len
selcol$conlen <- datlen$conlen
# core genome
# core if shared in all of the member of population
core_gen <- selcol[selcol$comcol == nosamp, "conlen"] %>% sum()
# flex genome if shared less than the total of population
# not consider node not present
if (nosamp == 1)
flex_gen <- 0
else
flex_gen <- selcol[selcol$comcol < nosamp & selcol$comcol > 0, "conlen"] %>% sum()
# tot_genome if at least observed in a single breeds
tot_gen <- selcol[selcol$comcol > 0, "conlen"] %>% sum()
datpan <- rbind(datpan, data.frame(
norep = norep,
nosamp = nosamp,
assemb = paste(selsamp, collapse = ","),
core_gen = core_gen,
flex_gen = flex_gen,
tot_gen = tot_gen
))
}
}
datpan
p1<-datpan %>%
select(nosamp,core_gen,tot_gen) %>%
mutate(nosamp=factor(nosamp)) %>%
pivot_longer(!nosamp) %>%
ggplot(aes(x=nosamp,y=value))+
geom_boxplot(aes(fill=name))+
geom_smooth(aes(x=as.numeric(nosamp),color=name))+
#geom_violin(aes(fill=name))+
theme_bw(base_size = 20)+
theme(panel.grid = element_blank(),
legend.position = "top",
legend.title = element_blank())+
scale_y_continuous(labels = function(x){paste(x/1000000,"M")})+
labs(x="Sample Number",y="Genome Size")+
scale_color_manual(values = c("#e3010a","#00b2ec"))+
scale_fill_manual(values = c("#e3010a","#00b2ec"))
p2<-datpan %>%
select(nosamp,core_gen,tot_gen) %>%
mutate(nosamp=factor(nosamp)) %>%
pivot_longer(!nosamp) %>%
ggplot(aes(x=nosamp,y=value,color=name))+
geom_jitter(width = 0.1,size=5,alpha=0.8)+
geom_smooth(aes(x=as.numeric(nosamp)))+
#geom_violin(aes(fill=name))+
theme_bw(base_size = 20)+
theme(panel.grid = element_blank(),
legend.position = "top",
legend.title = element_blank())+
scale_y_continuous(labels = function(x){paste(x/1000000,"M")})+
labs(x="Sample Number",y="Genome Size")+
scale_fill_manual(values = c("#4da0a0","#9b3a74"))+
scale_color_manual(values=c("#4da0a0","#9b3a74"))
library(patchwork)
p1+p2
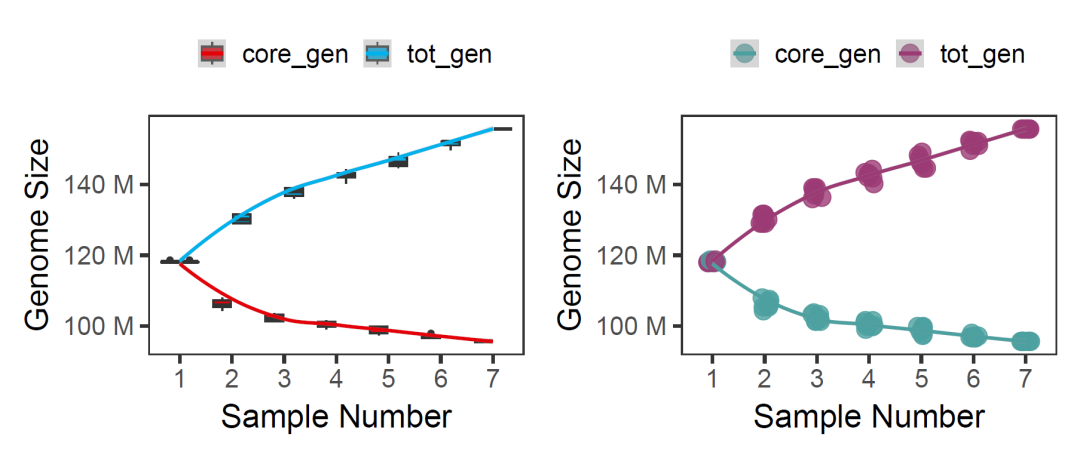
image.png
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-12-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号