全长转录组 | Oxford Nanopore (ONT) 三代全长转录组分析流程 -- 数据质控和预处理
原创
全长转录组 | Oxford Nanopore (ONT) 三代全长转录组分析流程 -- 数据质控和预处理
原创
三代测序说
修改于 2024-02-05 10:53:31
修改于 2024-02-05 10:53:31
ONT全长转录组测序是指基于牛津纳米孔公司(Oxford Nanopore Technologies,ONT)三代测序平台进行的全长转录组测序。利用三代测序平台长度长 (long-read)的特性,无需对转录本进行片段化,直接获取某一物种mRNA(或者有polyA尾的lncRNA)5'端到3'端的高质量全长转录组序列信息(图1),可准确识别可变剪接、基因融合、基因家族、可选择性多聚腺苷酸化 (alternative polyadenylation, APA)、等位基因特异性表达等转录本结构方面的变异。基于ONT三代测序平台进行全长转录组测序,除了可准确鉴别上述转录本结构变异,由于现阶段测序成本和通量(相对于PacBio平台),还可实现转录本(mRNA或polyA+ lncRNA)表达水平准确定量和差异分析。
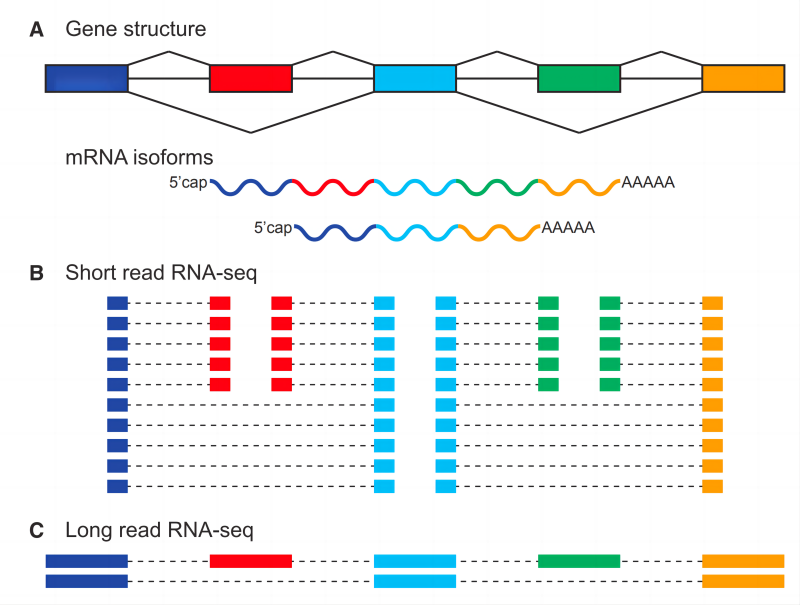
图1. 三代转录组测序之于二代测序的优势 Park, Eddie et al
一、ONT测序技术简介
ONT测序是基于电信号识别碱基序列的三代测序技术。DNA/RNA上不同碱基化学性质存在差异,单个核酸分子在分子马达的带领下与镶嵌在生物膜上的纳米孔蛋白结合并解旋,通过纳米孔通道时,碱基造成的阻碍大小不一,因此会形成特征性离子电流变化信号。通过对这些信号进行实时检测,即可获得相应碱基类型,完成测序 (图2)。目前通过“递归神经网络(Recurrent Neural Network)”的复杂算法对碱基进行判读。
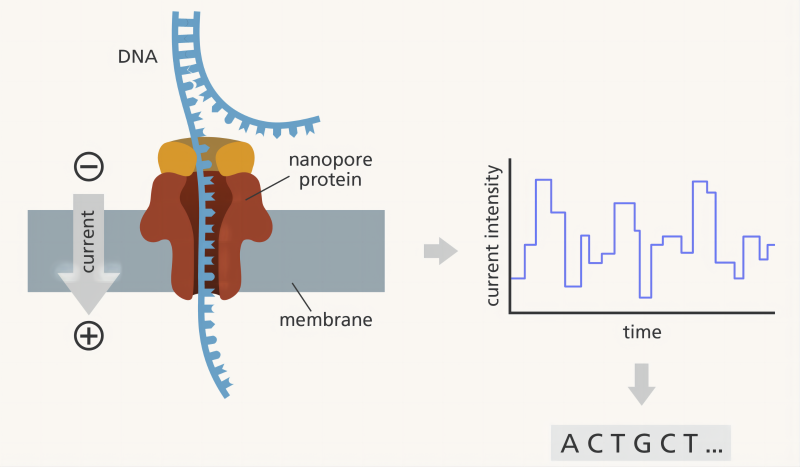
图2. ONT测序平台工作原理 Image credit: Laura Olivares Boldú, Wellcome Connecting Science
ONT测序技术具有以下特点: 1)读长长,最长读长能达到4.2 M以上级别,有利于可变剪接、基因融合等结构变异检测; 2)低成本:相比其他三代测序技术,ONT测序样本处理极其简单,无需DNA聚合酶、连接酶和dNTPs,测序价格低; 3)可不进行PCR扩增,避免二代测序中PCR扩增可能引入的错误或丰度变化; 4)RNA/DNA-direct方式建库,可直接读取碱基修饰信息,如甲基化修饰5mC、6mA等,无须像二代测序需要经过重硫酸盐转化或者免疫沉淀富集实验; 5)无GC含量和碱基偏好性,转录本表达定量准确。
二、ONT全长转录组测序实验流程
按照Oxford Nanopore Technologies(ONT)公司提供的cDNA-PCR(最新的为SQK-PCS114)建库文档执行,包括样品质量检测、文库构建、文库质量检测和文库测序等流程,以cDNA-PCR方式进行展示 (图3)。起始投入10ng 富集的RNA (polyA富集或者核糖体去除)或,500ng 总RNA。
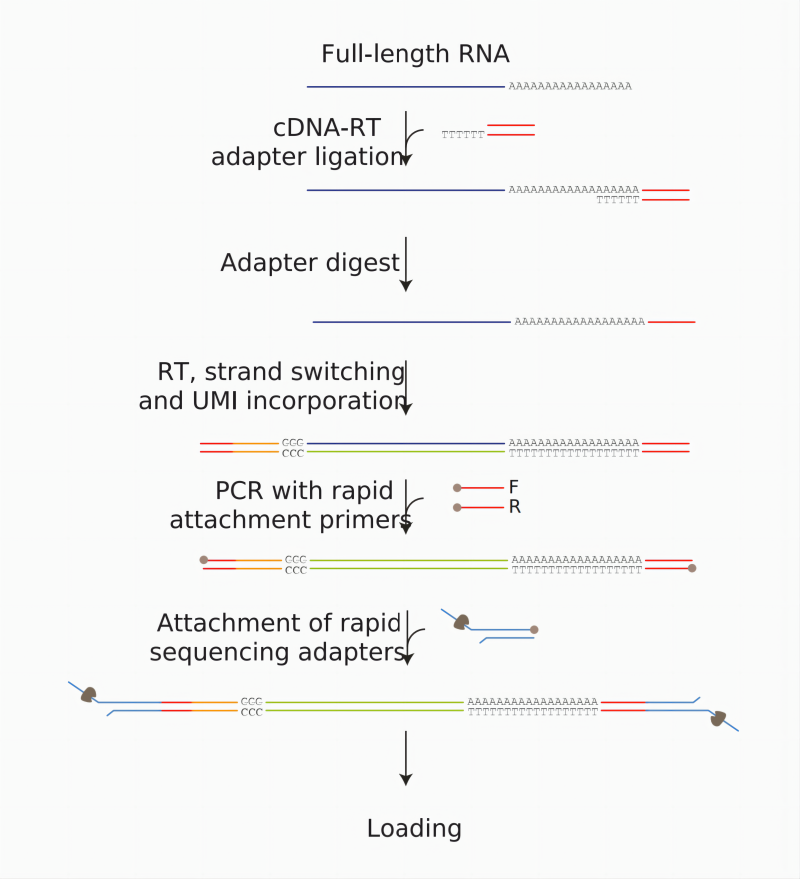
图3. cDNA-PCR全长转录组建库流程
三、ONT全长转录组的分析流程
PacBio全长转录组有官方自己开发优化的转录本聚类软件软件和流程,IsoSeq(https://isoseq.how/)。ONT全长转录组的分析更多的依赖于第三方开发的软件和流程,其基于minimap2和StringTie2搭建的 wf-transcriptomes流程部署在epi2me-labs里供用户使用。
现有还原重构全长转录本的分析软件大体分为两大类。一类是依赖参考基因组及其注释文件,如FLAIR,TAMA,TALON等;一类是reference-free,如IsoSeq, LyRic。还有就是老牌RNA-seq分析软件,StringTie2,可以结合short reads(二代测序数据)和 long reads(三代测序数据);IsoQuant 和 StringTie2既能依赖参考基因组注释,也能de novo annotation-free重构全长转录本。
ONT全长转录组数据分析前,需要对下机数据进行质控和全长转录本的鉴定,才能作为上述软件的输入文件,这次我们先对ONT数据进行质控和预处理,将数据准备好,进行下一步的分析。由于IsoQuant,Bambu,FLAIR,TAMA,TALON等软件既适用于ONT也适用于PacBio的全长转录组数据,因此后面我将会针对每一个软件的使用出一个教程。
ONT全长转录组演示数据来自Europen Nucleotide Archive(ENA)数据库:PRJEB31662,下载了ERR3218377.fastq.gz,ERR3218373.fastq.gz两个样本的数据作为演示。
1. 原始下机数据fast5 -- Dorado
如果是原始的fast5数据,需要通过使用Dorado(或GUPPY)软件将电信号转化为碱基序列。一般来说,测序服务商会把拆分好barcode的.bam 或/和 .fastq 文件给到用户,不需要用户自己做转换。
2. 原始下机数据质量统计 -- NanoComp
关于ONT下机数据的质量统计和可视化,可参考前面的详细教程:三代测序 - Oxford Nanopore (ONT) 数据分析 - 数据质控和过滤。这里直接使用NanoComp一步到位:
- 使用软件 NanoComp:https://github.com/wdecoster/nanocomp
- 软件安装
$ pip install NanoComp- 软件使用
#官方使用示例
$ NanoComp --bam alignment1.bam alignment2.bam alignment3.bam --outdir compare-runs
$ NanoComp --fastq reads1.fastq.gz reads2.fastq.gz reads3.fastq.gz reads4.fastq.gz --names run1 run2 run3 run4
#实际样本
$ nohup NanoComp -t 24 -f pdf \
--fastq 0_raw_fq/ERR3218373.fastq.gz 0_raw_fq/ERR3218377.fastq.gz \
--names ERR3218373 ERR3218377 \
-o NanoComp &
# -f 图片以pdf的格式输出,{'png'(default),'jpg','jpeg','webp','svg','pdf','eps','json'}
# -t 运行线程数
# -o, --outdir OUTDIR 输出文件文件夹如果测序数据质量,读长符合预期标准,则进行下一步。
3. 原始下机数据质控 -- Chopper
对下机原始序列进行过滤(按长度 或/和 平均碱基质量),剪切(序列头尾)和污染序列(--contam)的去除可以使用Chopper。
- 使用软件 chopper:https://github.com/wdecoster/chopper
- 软件安装
$ conda install -c bioconda chopper- 软件使用
#官方使用示例
$ gunzip -c reads.fastq.gz | chopper -q 10 -l 500 | gzip > filtered_reads.fastq.gz
#实际样本
$ gunzip -c 0_raw_fq/ERR3218373.fastq.gz | chopper -q 7 -l 50 | gzip > 1_chopper/ERR3218373.fastq.gz &
$ gunzip -c 0_raw_fq/ERR3218377.fastq.gz | chopper -q 7 -l 50 | gzip > 1_chopper/ERR3218377.fastq.gz &
#运行结果
Kept 291095 reads out of 525588 reads
Kept 267866 reads out of 604601 reads- 去除平均碱基质量小于7的reads,这个值一般是下机数据的预设值,所以如果想进行更加严格的质控,可以将这个数值设置的大一些。
- 去除reads长度小于50bp的序列,这个数值也可以自行调整。去除小片段reads和高度降解reads,减少非特异性比对。
- 如果测序过程中存在污染序列可以通过
--contam去除特定污染序列,比如支原体污染序列。
4. 全长转录本序列鉴定 -- Pychopper
Pychopper是鉴定,定向和修剪全长Nanopore cDNA序列的工具,该工具还可以修复融合的序列。
- 使用软件
Pychopper:https://github.com/epi2me-labs/pychopper
- 软件安装
$ conda install -c nanoporetech -c conda-forge -c bioconda "nanoporetech::pychopper"- 软件使用
#官方使用示例
#Example usage with default PCS109/DCS109 primers using the default pHMM backend.
$ pychopper -r report.pdf -u unclassified.fq -w rescued.fq input.fq full_length_output.fq
#实际样本
$ mkdir 2_pychopper
$ nohup pychopper -t 8 -r 2_pychopper/ERR3218373_report.pdf \
-u 2_pychopper/ERR3218373_unclassified.fq \
-w 2_pychopper/ERR3218373_rescued.fq \
1_chopper/ERR3218373.fastq.gz 2_pychopper/ERR3218373_full_length.fq \
1> 2_pychopper/ERR3218373.pychopper.log 2>&1 &
$ nohup pychopper -t 8 -r 2_pychopper/ERR3218377_report.pdf \
-u 2_pychopper/ERR3218377_unclassified.fq \
-w 2_pychopper/ERR3218377_rescued.fq \
1_chopper/ERR3218377.fastq.gz 2_pychopper/ERR3218377_full_length.fq \
1> 2_pychopper/ERR3218377.pychopper.log 2>&1 &
# -r report_pdf Report PDF (pychopper_report.pdf).
# -u unclass_output Write unclassified reads to this file
# -w rescue_output Write rescued reads to this file.
# -Q min_qual Minimum mean base quality (7.0).
# -z min_len Minimum segment length (50).pychopper只有检测到双端的引物才能识别定义为全长完整(full length)的序列,所以在这之前不要对序列两端的引物(primers)进行裁剪去除,包括在用Dorado电信号转化碱基的时候。- 通过
-Q和-z可以实现chopper的功能,所以如果没有污染序列去除的话,可以跳过第二部直接使用pychopper。 - -k 可以指定试剂盒和protocol。kit{PCS109,PCS110,PCS111,LSK114}。
- 最后得到的全长转录本序列
full_lenght.fastq可用于后续的分析。
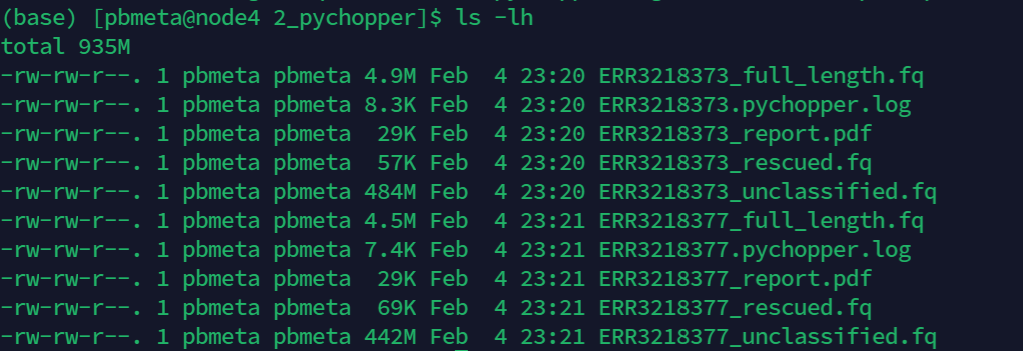
图4. pychopper结果文件
参考文献:
- Nanopore三代全长转录组
- ONT全长转录组测序系列一:初识篇
- 基因结构预测新利器-ONT全长转录组
- Park, Eddie et al. "The expanding landscape of alternative splicing variation in human populations." The American Journal of Human Genetics (2018).
- Mestre-Tomás, Jorge, et al. "SQANTI-SIM: a simulator of controlled transcript novelty for lrRNA-seq benchmark." Genome Biology 24.1 (2023): 286.
- Nanopore全长转录组测序
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号