seurat v5直播,一键完成五种数据整合:harmony,CCA,RPCA,FastMNN,scVI
原创
seurat v5直播,一键完成五种数据整合:harmony,CCA,RPCA,FastMNN,scVI
原创
生信小博士
发布于 2024-02-23 23:16:54
发布于 2024-02-23 23:16:54
龙年大吉! 很高兴在正月初一与大家相见。
本文主要测试:seuratv5环境下,五种单细胞整合方法
- · CCA方法整合
- · RPCA方法整合
- · Harmony方法整合
- · FastMNN方法整合
- · scVI方法整合

生信小博士
【生物信息学】R语言,学习生信,seurat,单细胞测序,空间转录组。 Python,scanpy,cell2location,资料分享
- 首先在v5环境下,加载pbmc
2 #https://satijalab.org/seurat/articles/install_v5.html#2在seurat_v5文件夹下安装v5---.libPaths( c( '/home/rootyll/seurat_v5/', "/usr/local/lib/R/site-library", "/usr/lib/R/site-library", "/usr/lib/R/library" ))
library(Seurat)pbmc = readRDS('~/gzh/pbmc3k_final.rds')
DimPlot(pbmc)- 我们能看到此时的pbmc对象还是一个seurat v4对象

- 第二步:把seuratv4对象转为seuratv5对象,实现seuratv4对象和v5对象共存:使用seuratv5 读取seurat v4制作的rds文件
pbmc[["RNA5"]] <- as(object = pbmc[["RNA"]], Class = "Assay5")
DefaultAssay(pbmc)Assays(pbmc)
pbmc[["RNA_seuratv4"]] <- pbmc[['RNA']]
pbmc[['RNA']]=pbmc[['RNA5']]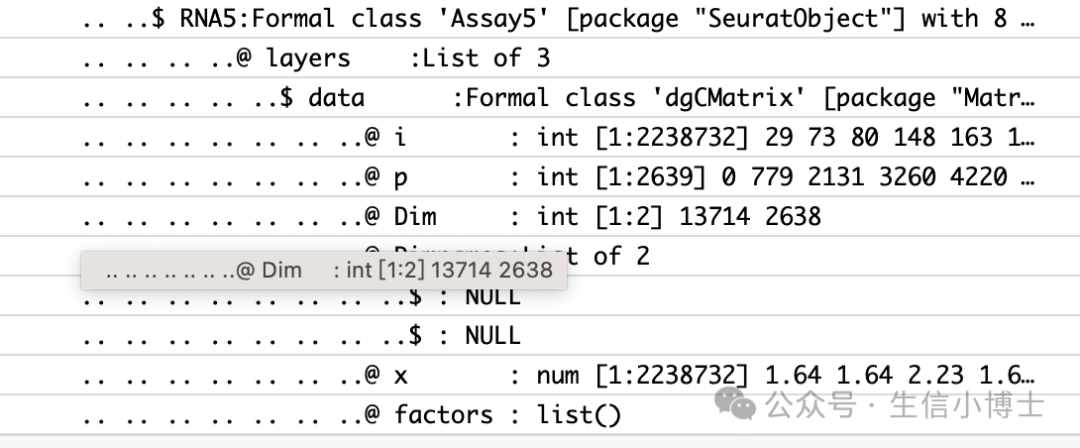
- 如果是单个样本,走下面的seurat标准流程即可。v4和v5的标准流程没啥区别
#3 v5对象标准流程----#向pbmc新增一列percent.mt数据
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
#使用小提琴图可视化QC指标
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
#FeatureScatter通常用于可视化 feature-feature 相关性,#nCount_RNA 与percent.mt的相关性
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
#nCount_RNA与nFeature_RNA的相关性
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2 #合并两图
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
#选取 2500 > nFeature_RNA >200 和percent.mt < 5的数据
pbmc <- NormalizeData(object = pbmc)pbmc <- FindVariableFeatures(object = pbmc)
pbmc <- ScaleData(object = pbmc)
pbmc <- RunPCA(object = pbmc)
pbmc <- FindNeighbors(object = pbmc, dims = 1:30)
pbmc <- FindClusters(object = pbmc)
pbmc <- RunUMAP(object = pbmc, dims = 1:30)
DimPlot(object = pbmc, reduction = "umap")结果如下
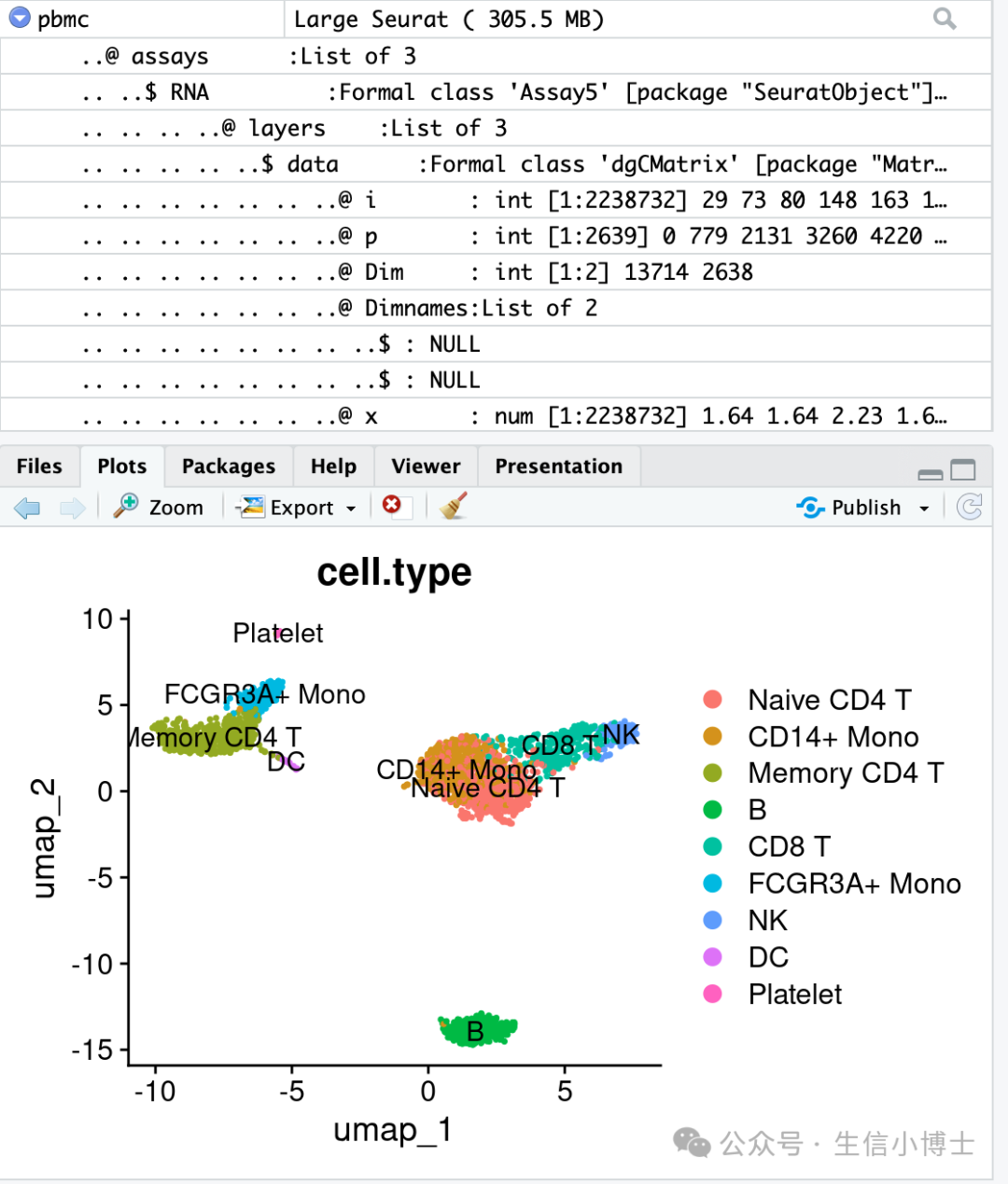
- 如果是多个样本处理的话,就需要进行样本整合了。seuratv5版本集合了5种整合方法,我们来尝试一下:
- 首先,我们需要创建两个seuratv5对象
#为了演示,我们把pbmc对象分成2个数据集,进行整合分析
dim(pbmc)#13714 2638
pbmc$group= ifelse(pbmc$nCount_RNA>2200,yes = "CTRL",no = 'STIM')
table(pbmc$group)4##4 # In line with prior workflows, you can also into split your object into a list of multiple objects based on a metadata# column creates a list of two objects
ifnb_list <- SplitObject(pbmc, split.by = "group")
ifnb_list$CTRLifnb_list$STIM
- 得到两个seuart对象之后,下面进行整合分析
- · CCA方法整合
- · RPCA方法整合
- · Harmony方法整合
- · FastMNN方法整合
- · scVI方法整合
- 时间所限,这里只进行了cca、rpca和harmony的整合方法
#4我们有两个seuratv5的对象,进行整合分析------merged_obj <- merge(x = ifnb_list$CTRL, y = ifnb_list$STIM)merged_obj <- NormalizeData(merged_obj)merged_obj <- FindVariableFeatures(merged_obj)merged_obj <- ScaleData(merged_obj)merged_obj <- RunPCA(merged_obj)obj=merged_obj#rpcaobj <- IntegrateLayers(object = obj, method = RPCAIntegration, orig.reduction = "pca", new.reduction = "integrated.rpca", verbose = FALSE)#ccaobj <- IntegrateLayers( object = obj, method = CCAIntegration, orig.reduction = "pca", new.reduction = "integrated.cca", verbose = FALSE)#remotes::install_github("satijalab/seurat-wrappers")#BiocManager::install('batchelor')#BiocManager::install('SeuratData',force = TRUE)# obj <- IntegrateLayers(# object = obj, method = FastMNNIntegration,# new.reduction = "integrated.mnn",# verbose = FALSE# )# # SeuratWrappers::RunFastMNN(object.list = obj,reduction.name = 'mnn')#harmonyobj <- IntegrateLayers( object = obj, method = HarmonyIntegration, orig.reduction = "pca", new.reduction = "harmony", verbose = FALSE)- 这里只展示cca和harmony的整合结果
obj <- FindNeighbors(obj, reduction = "integrated.cca", dims = 1:30)
obj <- FindClusters(obj, resolution = 2, cluster.name = "cca_clusters")
obj <- RunUMAP(obj, reduction = "integrated.cca", dims = 1:30, reduction.name = "umap.cca")
p1 <- DimPlot( obj, reduction = "umap.cca", group.by = c("Method", "predicted.celltype.l2", "cca_clusters"), combine = FALSE, label.size = 2)
obj <- FindNeighbors(obj, reduction = "harmony", dims = 1:30)obj <- FindClusters(obj, resolution = 2, cluster.name = "harmony_clusters")
obj <- RunUMAP(obj, reduction = "harmony", dims = 1:30, reduction.name = "harmony")
p2 <- DimPlot( obj, reduction = "harmony", group.by = c("Method", "cell.type", "harmony_clusters"), combine = FALSE, label.size = 2)
library(patchwork)
wrap_plots(c(p1, p2), ncol = 2, byrow = F)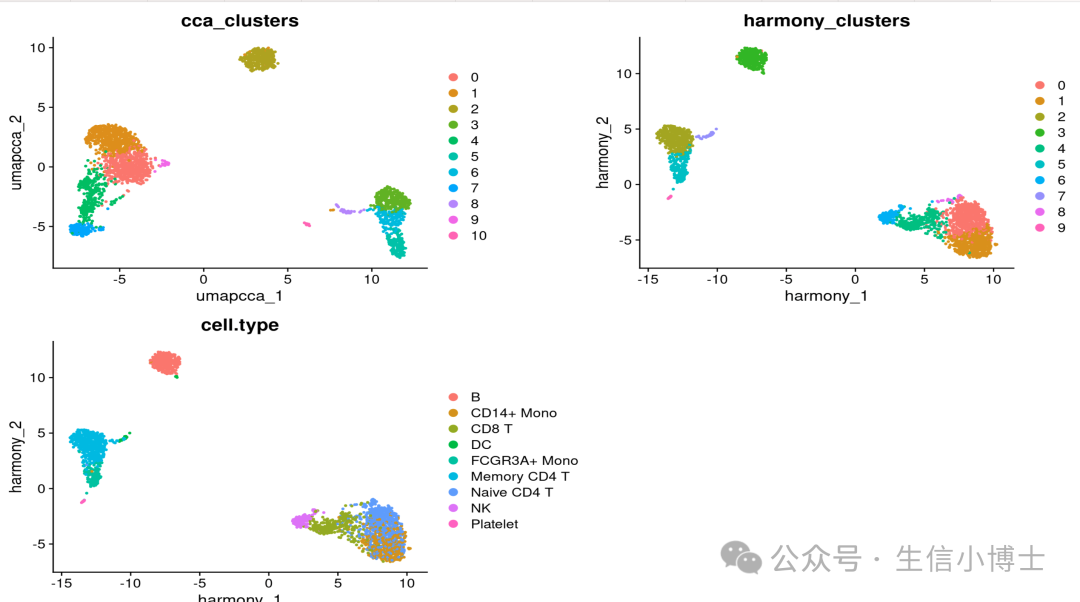
看上去一致性还可以,但是后续可以直播本篇推文的5种方法,看看哪种方法又快又好。
参考:
- seuratv5标准流程:https://satijalab.org/seurat/articles/essential_commands
- seuratv4和seruatv5的区别:https://satijalab.org/seurat/articles/announcements.html
- seratv5整合分析:https://satijalab.org/seurat/articles/seurat5_integration https://satijalab.org/seurat/articles/integration_introduction
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号