scanpy和seurat的所有Marker基因可视化方法帮你打包好啦

scanpy和seurat的所有Marker基因可视化方法帮你打包好啦

生信技能树jimmy
发布于 2024-03-21 12:11:49
发布于 2024-03-21 12:11:49
我们在进行单细胞亚群命名时,是通过Marker基因来确定细胞的身份。然而在注释过程中,Marker基因的可视化是必不可少的,以前我们做了一个投票:可视化单细胞亚群的标记基因的5个方法,是基于R编程语言的Seurat包的5个基础函数相信大家都是已经烂熟于心了:
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
- FeaturePlot(pbmc, features = c("MS4A1", "CD79A"))
- RidgePlot(pbmc, features = c("MS4A1", "CD79A"), ncol = 1)
- DotPlot(pbmc, features = unique(features)) + RotatedAxis()
- DoHeatmap(subset(pbmc, downsample = 100), features = features, size = 3)
接下来我们一起看看基于R编程语言的Seurat包的5个基础函数的可视化,如何使用Python编程语言进行“平替”:
基于R编程语言的Seurat包
library(Seurat)
library(ggplot2)
library(gridExtra)
#加载实例数据
data('pbmc_small')
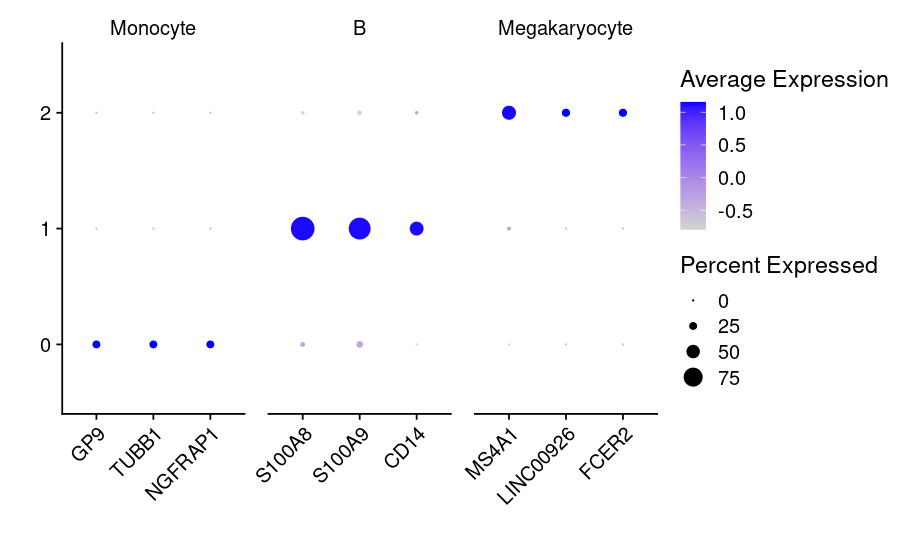
1.dotplot可视化
DotPlot(pbmc_small,features=list(Monocyte=c("GP9","TUBB1","NGFRAP1"),
B=c("S100A8","S100A9","CD14" ),Megakaryocyte=c("MS4A1","LINC00926","FCER2" )))+
RotatedAxis()+labs(x='',y='')
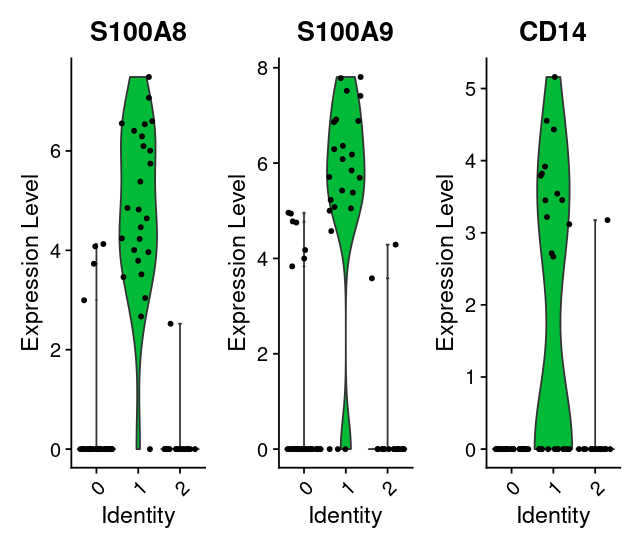
2.violin plot
VlnPlot(pbmc_small,group.by ='RNA_snn_res.1',features = c("S100A8","S100A9","CD14"))
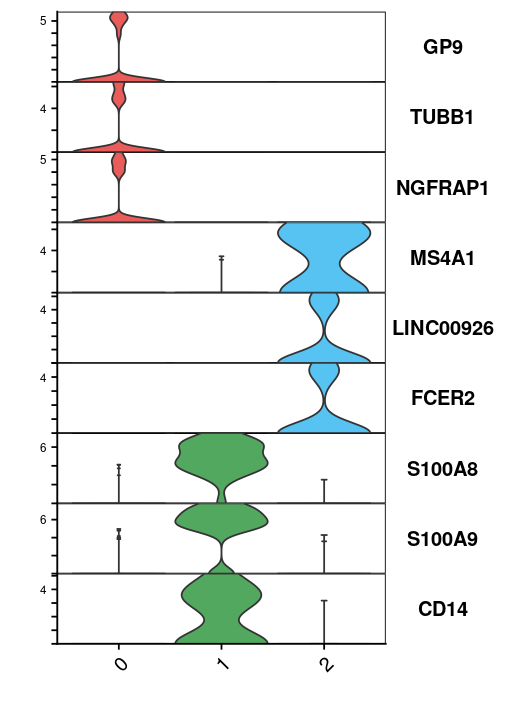
3.stacked-violin plot
VlnPlot(pbmc_small,
features = c("GP9","TUBB1","NGFRAP1",
"MS4A1","LINC00926","FCER2",
"S100A8","S100A9","CD14"),
split.by = 'RNA_snn_res.1',stack = TRUE,
flip=TRUE,
cols=c('#E95C59', '#53A85F', '#57C3F3'))+
theme(legend.position='none')+labs(x='',y='')
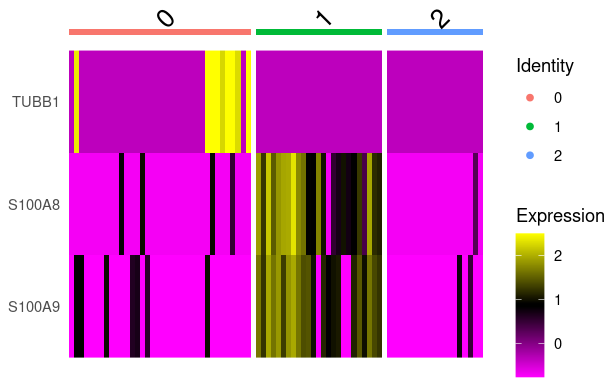
4.Heatmap
DoHeatmap(pbmc_small,features = c("TUBB1","S100A8","S100A9"))
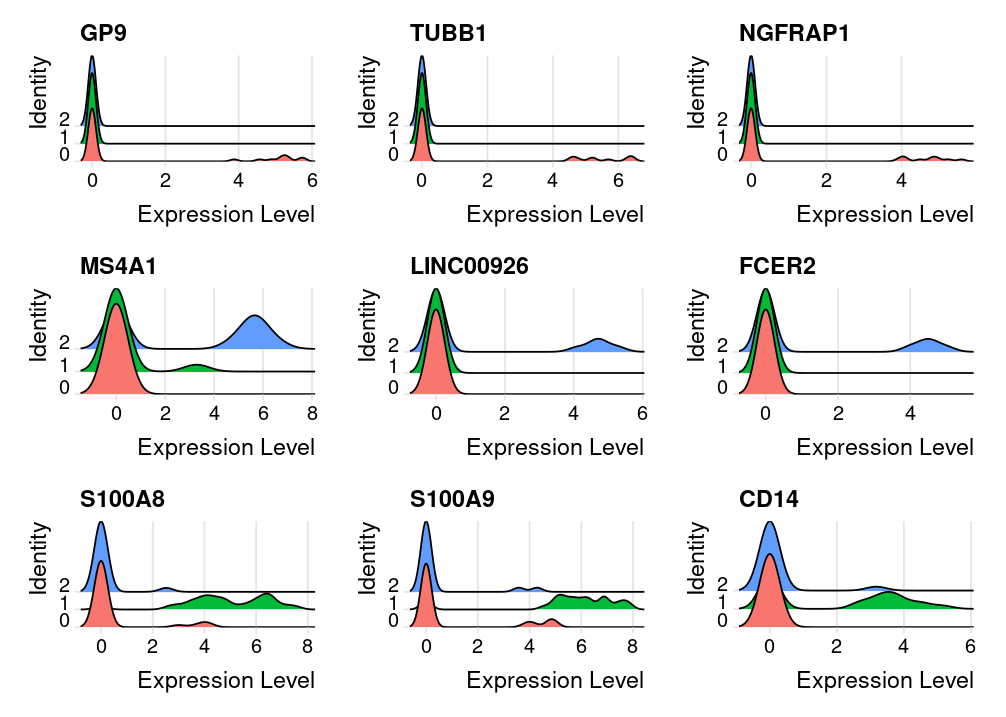
5.RidgePlot
原版:
RidgePlot(pbmc_small, features = c("GP9","TUBB1","NGFRAP1",
"MS4A1","LINC00926","FCER2",
"S100A8","S100A9","CD14"))
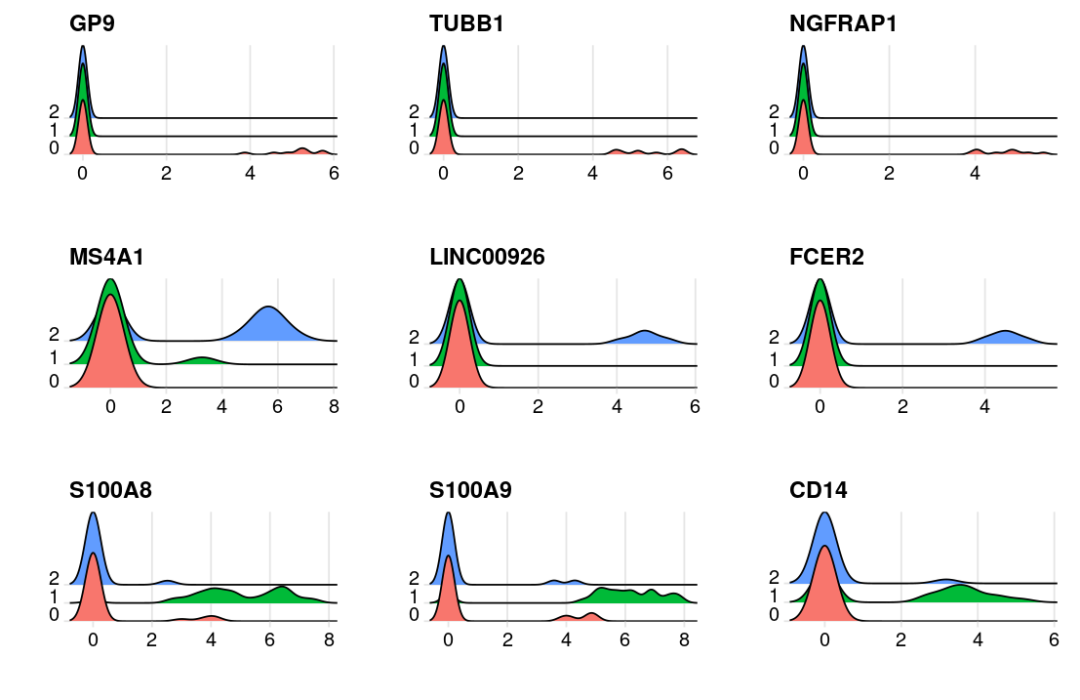
个性版:
genes= c("GP9","TUBB1","NGFRAP1","MS4A1","LINC00926","FCER2","S100A8","S100A9","CD14")
pList = lapply(genes, function(x){
RidgePlot(pbmc_small,features =x )+labs(x='',y='')+NoLegend()
})
gridExtra::grid.arrange(grobs = pList, ncol = 3)
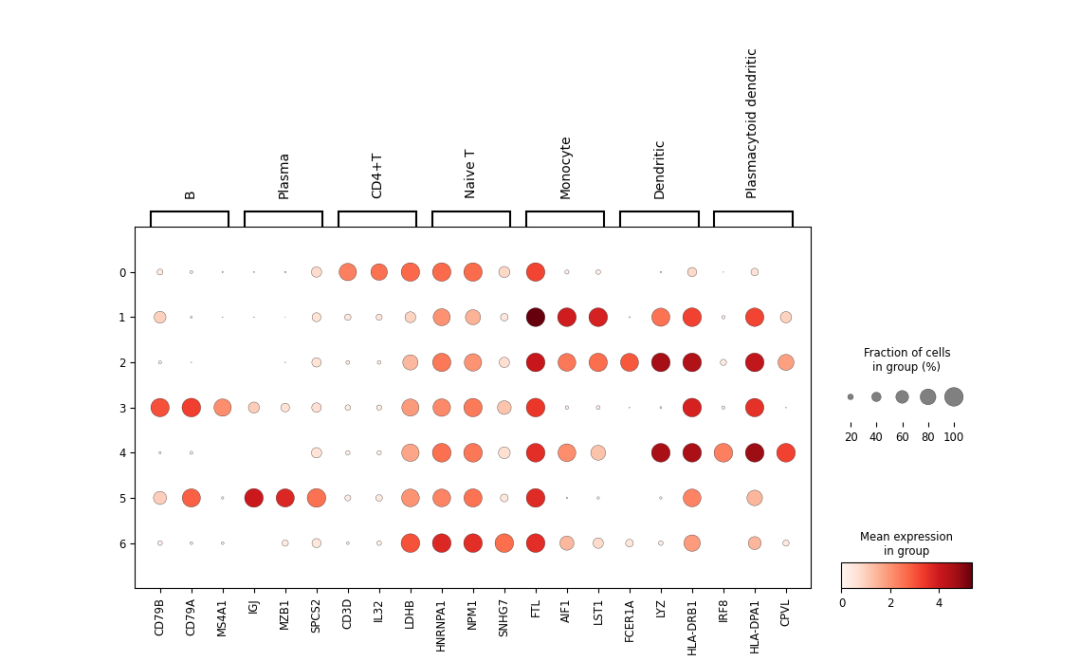
scanpy
import scanpy as sc
#加载实例数据
adata=sc.datasets.pbmc68k_reduced()
sc.tl.leiden(adata,resolution=0.2)
markers={'B':['CD79B','CD79A','MS4A1'],
'Plasma':['IGJ','MZB1','SPCS2'],
'CD4+T':['CD3D','IL32','LDHB'],
'Naive T':['HNRNPA1','NPM1','SNHG7'],
'Monocyte':['FTL','AIF1','LST1'],
'Dendritic':['FCER1A','LYZ','HLA-DRB1'],
'Plasmacytoid dendritic':['IRF8','HLA-DPA1','CPVL'],
}
1.dotplot可视化
sc.pl.dotplot(adata,markers,'leiden')
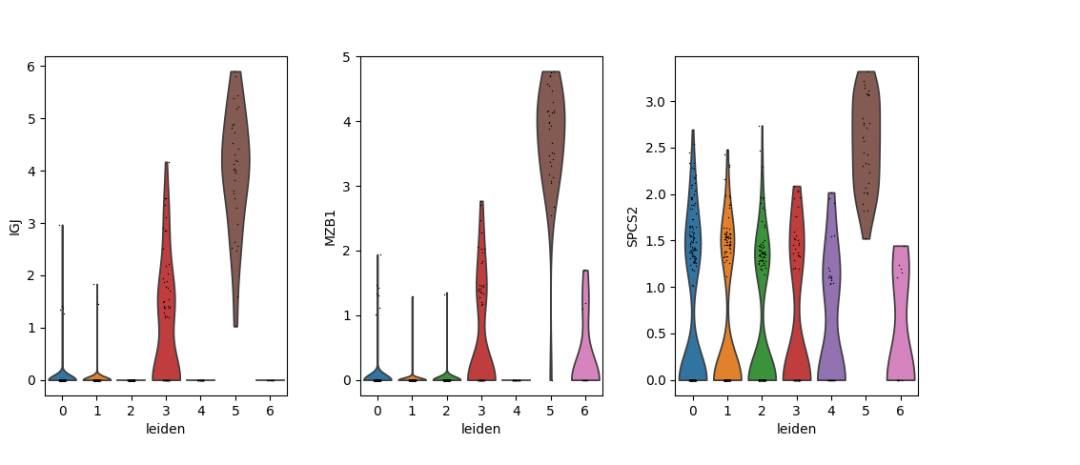
2.violin plot
sc.pl.violin(adata,['IGJ','MZB1','SPCS2'],'leiden')
3.stacked-violin plot
sc.pl.stacked_violin(adata,markers,'leiden')
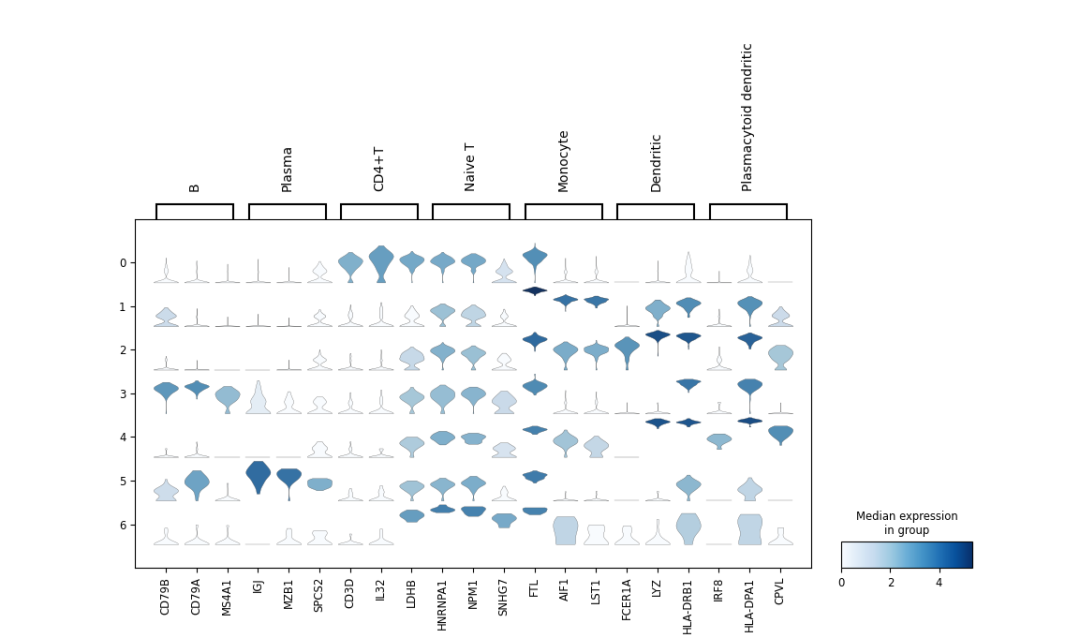
4.matrixplot
sc.pl.matrixplot(adata,markers,'leiden',standard_scale='var')
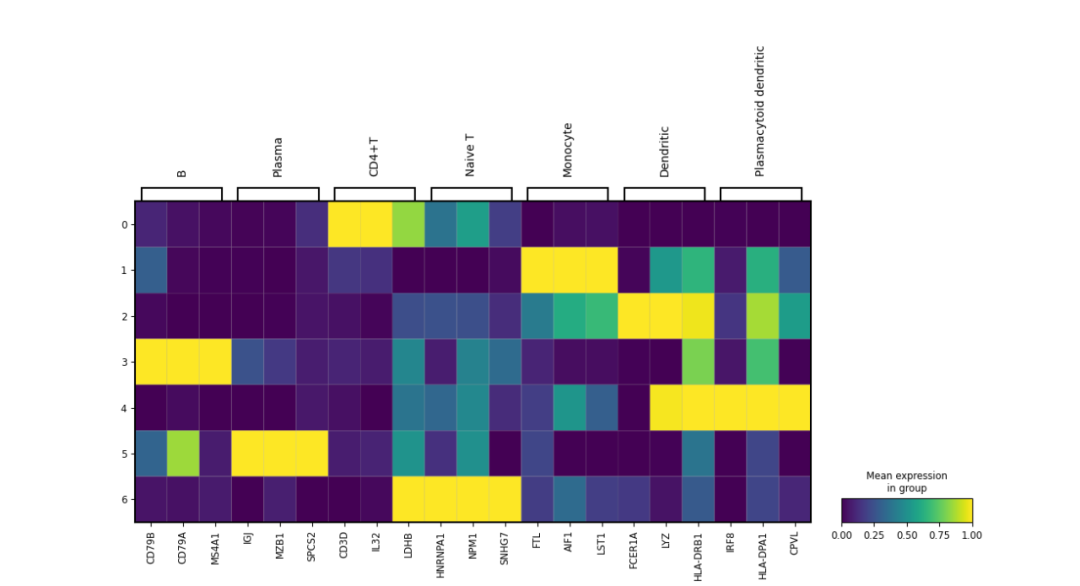
值得注意的,这个matrixplot可视化方法在基于R编程语言的Seurat包的里面没有对应的方法哦!其实这个matrixplot可视化方法就是下面的这个heatmap可视化方法的亚群平均值。
5.heatmap
sc.pl.heatmap(adata,markers,'leiden',var_group_rotation=True)
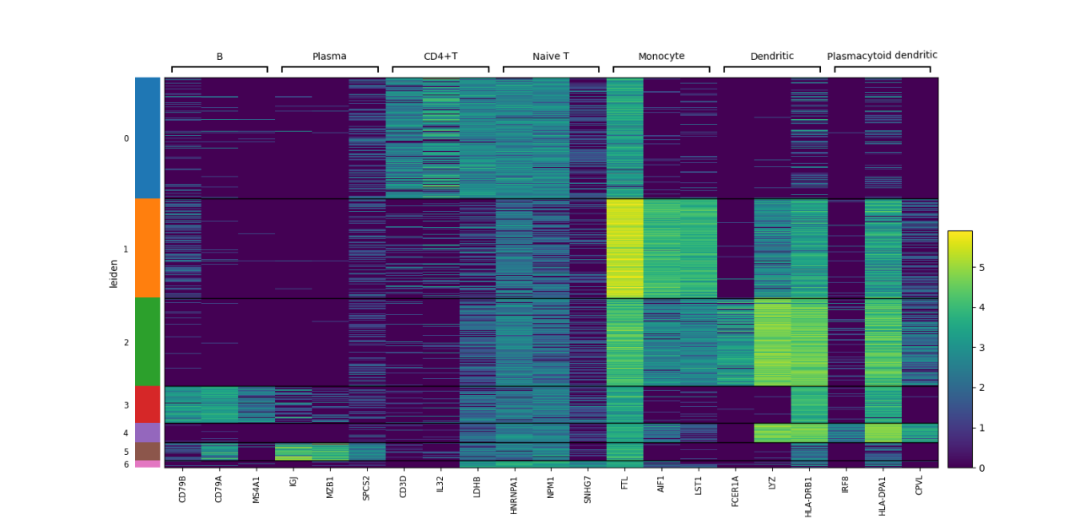
6.tracksplot
sc.pl.tracksplot(adata,markers,'leiden')
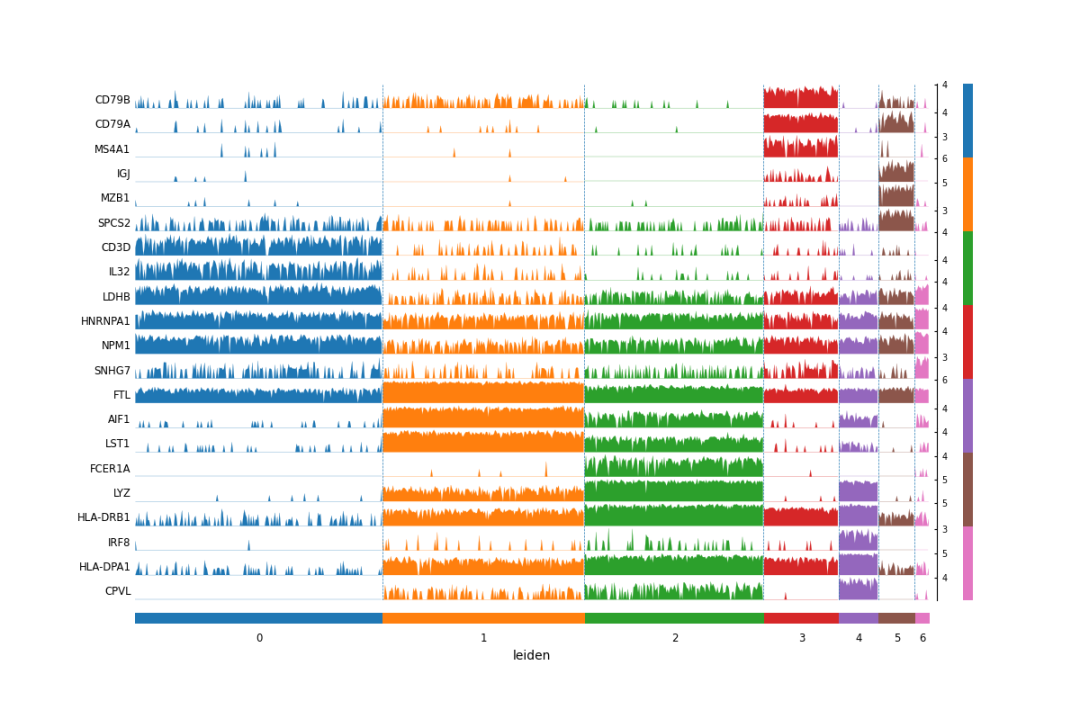
上面的是通过已知的基因标记来确定各个单细胞亚群的特征并且可视化,所以是需要自己准备好基因集,那些有生物学意义的基因。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-03-20,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号