全长转录组 | 三代全长转录之circRNA(ONT )-- CIRI-long
原创
全长转录组 | 三代全长转录之circRNA(ONT )-- CIRI-long
原创
三代测序说
修改于 2024-04-07 16:44:55
修改于 2024-04-07 16:44:55
环状RNA(circular RNA,circRNA)是一类特殊的非编码RNA(noncoding RNA,ncRNA),也是RNA领域最新的研究热点。与传统的线性RNA(linear RNA,含5’和3’末端)不同,circRNA分子呈封闭环状结构,不受RNA外切酶影响,表达更稳定,不易降解。
目前研究表明,在生物体内,circRNA主要通过其序列特征,发挥miRNA海绵、RNA-binding proteins (RBPs)海绵以及翻译短肽等生物学功能(1-2)。因此,确定其的全长序列,是进行circRNA功能研究的重要基础。由于目前对于circRNA的研究多采用二代测序的方法,而circRNA的内部序列与线性mRNA分子高度相似,单纯通过算法(识别反向剪切位点)很难区分来自环形RNA和线性RNA分子的读段,以及确定全长circRNA内部组成。近期的研究中利用了长读长测序技术,对circRNA的全长重构进行了尝试(3-4)。因此,目前研究方法对于circRNA结构的识别能力主要被二代测序的读长所限制,对于长度较长(>500bp)的circRNA分子,仍然缺少有效的全长重构手段。
赵方庆教授团队前期提出了CIRI-AS算法(基于BSJ读段对比结果对环形RNA内部可变剪接结构进行识别)。后续研究开发了CIRI-full算法(通过识别双端250bp测序数据中反向重叠区特征,对500bp以内的环形RNA进行全长重构)。上述方法主要基于短读长测序技术,难以对长度500bp以上的circRNA的全长序列进行有效识别。
在此基础上,2021年3月11日,中国科学院北京生命科学研究院赵方庆教授团队在Nature Biotechnology杂志上发表了题为Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long 的文章,开发了一种基于三代纳米孔测序平台(Oxford Nanopore Technologies ,ONT)高效测定circRNA全长转录本的实验和计算方法:利用随机引物对circRNA进行的滚环反转录扩增后,使用三代纳米孔测序技术(ONT)对circRNA的全长序列进行直接测序,并开发了CIRI-long 算法,实现对长测序读段中的circRNA序列进行识别和全长重构。实验结果表明,与传统的circRNA二代测序技术相比,该方法将circRNA检测灵敏度提升了20倍,并可实现对不同长度(<100bp - 5kb)的circRNA全长序列的无偏识别,大幅提升了环形转录本的重构能力,为其功能研究提供了重要的实验方法和计算工具。
赵方庆教授实验室主页(图1):https://bioinfo.biols.ac.cn/
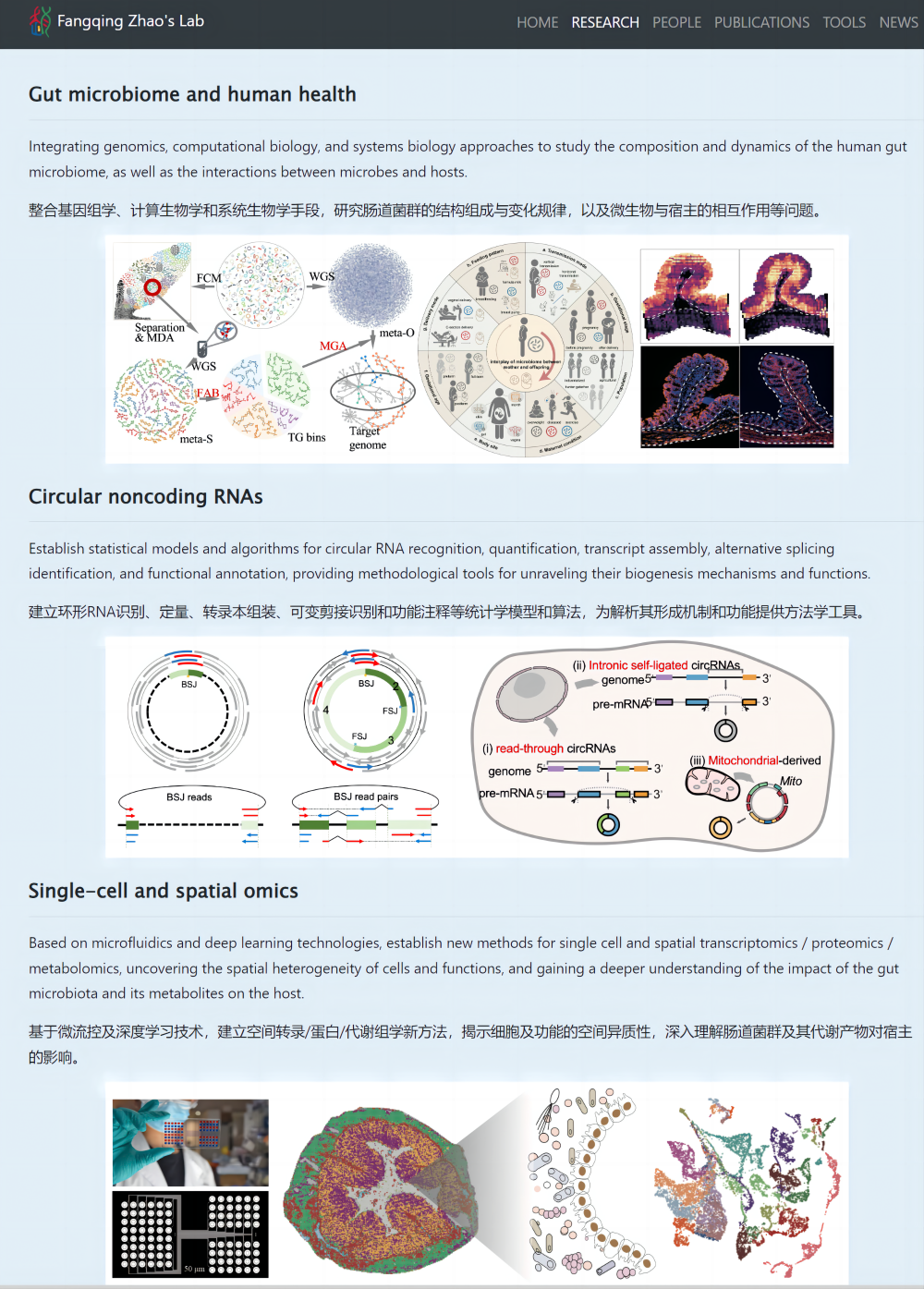
图1.赵方庆教授实验室主页
一、CIRI-long软件介绍
因为circRNAs及其对应的线性信使RNA之间的相似性,利用短读长RNA测序重建circRNA的全长序列一直是具有挑战性的,先前的测序方法无法实现对全长circRNA的高通量检测。赵方庆教授团队开发了一种利用三代纳米孔(ONT)测序技术进行circRNA及其相应的异构体(isoform)富集和全长测序的方案。环状逆转录和片段大小选择能比先前方法从总RNA中多富集出20倍的circRNAs。我们开发了一个使用长度长测序数据(CIRI-long)circRNA鉴定软件,用于重建circRNAs的序列。该算法工作流程利用模拟数据,通过与 Illumina 测序以及定量实时RT-PCR 的比较进行了验证。作者使用CIRI-long来分析成年小鼠脑组织样本,并系统地对circRNAs进行注释分析,包括来自线粒体circRNAs。作者鉴定了一种新的内含子自连接circRNA的特殊的剪接和表达模式。此方法利用了三代纳米孔测序的长读长优势,实现了对全长circRNA序列的无偏重建(图2)。

图2. CIRI-long文章和摘要
二、CIRI-long的安装
依赖软件:
gcc 4.8+或clang 3.4+cmake 3.2+python>=3.7samtools=1.9或更高minimap2
1. 从源代码安装
$ git clone https://github.com/bioinfo-biols/CIRI-long.git CIRI-long
$ cd CIRI-long
# Create virtual environment
$ python3 -m venv venv
# Activate virtualenv
$ source ./venv/bin/activate
# Install CIRI-long
$ make
# Test for installation
$ make test2. 使用pip安装
个人推荐使用,方便快捷。
$ pip install CIRI-long三、CIRI-long的使用方法
软件主页:https://github.com/bioinfo-biols/CIRI-long
1. 基本用法
CIRI-long两个命令: CIRI-long call 和 CIRI-long collapse,因此整个流程分为两步。
usage: CIRI-long [-h] [-v] {call,collapse} ...
positional arguments:
{call,collapse} commands
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit2. 步骤1:circRNA 鉴定
- 基本用法
#主命令
$ CIRI-long call [-h] [-i READS] [-o DIR] [-r REF] [-p PREFIX] [-a GTF] [--canonical] [-t INT] [--debug]
optional arguments:
-h, --help show this help message and exit #帮助文档
-i READS, --in READS Input reads.fq.gz #输入文件
-o DIR, --out DIR Output directory, default: ./ #输出文件夹路径
-r REF, --ref REF Reference genome FASTA file #参考基因组ref.fa文件,需要用bwa进行索引
-p PREFIX, --prefix PREFIX
Output sample prefix, (default: CIRI-long) #输出文件前缀
-a GTF, --anno GTF Genome reference gtf, (optional) #基因组注释文件(可选)
-c CIRC, --circ CIRC Additional circRNA annotation in bed/gtf format,
(optional) #以bed/gtf格式输出circRNA注释文件(可选)
-t INT, --threads INT Number of threads, (default: use all cores) #线程数
--debug Run in debugging mode, (default: False) #纠错模式运行注意:
参考基因组需要bwa的索引。在运行CIRI-long之前,使用bwa index命令对参考基因组ref.fa文件进行索引。
- 使用示例
#下载演示数据
$ wget https://github.com/bioinfo-biols/CIRI-long/releases/download/v0.6-alpha/CIRI-long_test_data.tar.gz
#演示数据解压
$ tar zxvf CIRI-long_test_data.tar.gz
$ cd test_data
#使用```bwa index```命令对参考基因组文件进行索引
$ bwa index -a bwtsw mm10_chr12.fa mm10_chr12.fa
#运行CIRI-long鉴定circRNA
$ CIRI-long call -i test_reads.fa \ #输入文件
-o ./test_call \ #输出路径
-r mm10_chr12.fa \ #参考基因组
-p test \ #输出文件前缀
-a mm10_chr12.gtf \ #基因组注释文件
-t 8 #使用线程数- 输出文件
test_call
├── test.cand_circ.fa # 主要文件,circRNA序列文件。
├── test.json
├── test.log
├── test.low_confidence.fa # circRNA序列文件,低置信度。
└── tmp
├── ss.idx
├── test.ccs.fa
└── test.raw.fa
# 如果不加 -c 选项,则产生一个文件夹,7个文件- 使用非经典剪切信号
如果想使用其它剪切信号,可以在脚本
align.py修改SPLICE_SIGNAL,格式为:{(5’SS, 3’SS): Priority} 。
默认:
SPLICE_SIGNAL = {
('GT', 'AG'): 0, # U2-type
('GC', 'AG'): 1, # U2-type
('AT', 'AC'): 2, # U12-type
('GT', 'AC'): 2, # U12-type
('AT', 'AG'): 2, # U12-type
}3. 步骤2:isoform合并(collapose)
- 基本用法
可以将多个样本的circRNA结果合并。
#主命令
$ CIRI-long collapse [-h] [-i LIST] [-o DIR] [-p PREFIX] [-r REF] [-a GTF] [--canonical] [-t INT] [--debug]
optional arguments:
-h, --help show this help message and exit #帮助文档
-i LIST, --in LIST Input list of CIRI-long results #样本名称和路径的list文件
-o DIR, --out DIR Output directory, default: ./ #输出文件夹路径
-p PREFIX, --prefix PREFIX
Output sample prefix, (default: CIRI-long) #输出文件前缀
-r REF, --ref REF Reference genome FASTA file #参考基因组文件
-a GTF, --anno GTF Genome reference gtf, (optional) #参考基因组注释文件
-c CIRC, --circ CIRC Additional circRNA annotation in bed/gtf format,
(optional) #以bed/gtf格式输出circRNA注释文件(可选)
-t INT, --threads INT
Number of threads, (default: use all cores) #线程数
--debug Run in debugging mode, (default: False) #纠错模式运行需要先创建一个想要合并样本(*.cand_circ.fa)的名称和路径的list文本文件,以空格分隔。
#list 文件内容
sample1_name /path/to/sample1/cand_circ.fa
sample2_name /path/to/sample2/cand_circ.fa- 使用示例
创建一个名为test.list文本文件:
test ./test_call/test.cand_circ.fa运行CIRI-long collapse合并一个或多个样本结果。
$ CIRI-long collapse -i ./test.lst \ #输入文件
-o ./test_collpase \ #输出文件夹路径
-p test \ #文件前缀
-r ./mm10_chr12.fa \ #参考基因组
-a ./mm10_chr12.gtf \ #参考基因组注释文件
-t 8 #线程- 输出文件
test_collpase
├── test_collpase.expression
├── test_collpase.isoforms
├── test_collpase.info
├── test_collpase.log
├── test_collpase.reads
└── tmp
├── ss.idx
└── test_collpase.corrected.pkl
# 如果不加 -c 选项,则产生一个文件夹,6个文件- 输出文件格式
1)主要输出文件,GTF格式文件(test_collpase.info),包含所有circRNA的详细信息和circRNA反向剪切区域的注释列。
列 | 名称 | 描述 |
|---|---|---|
1 | chrom 染色体位置 | chromosome/contig name ---- 染色体或contig名称 |
2 | source 来源 | CIRI-long |
3 | type 类型 | circRNA |
4 | start 起始 | 5' back-spliced junction site ---- 5'端反向剪切位点 |
5 | end 结束 | 3' back-spliced junction site ---- 3'端反向剪切位点 |
6 | score 得分 | Number of total supported reads ---- 支持reads数 |
7 | strand 链 | strand information ---- 链信息 |
8 | . | . |
9 | attributes 特性 | attributes seperated by semicolon ---- 分号分隔的属性 |
属性列包含了几个预先定义的关键词及其赋值:
key关键词 | description描述 |
|---|---|
circ_id circRNA的ID | name of circRNA ---- circRNA名称 |
splice_site 剪切位点 | splicing signal of candidate circRNAs and numbers indicating shifted bases of aligned and annotated splice site. (e.g. AG-GT | 0-5) 候选circRNA剪切信号和实际剪切位点和注释的偏差碱基数 |
equivalent_seq 等同序列 | equivalent sequence of splice site ---- 同一个剪切位点对应的其它circRNA序列 |
circ_type circ类型 | circRNA types: exon/intron/intergenic ---- circRNA类型:外显子/内含子/基因间区 |
circ_len circ长度 | length of the major isoform of circRNA ---- circRNA主要异构体的长度 |
isoform 异构体 | structure of isoforms, isoforms are seperated by "|" and circular exons are seperated by "," (e.g. 11627815-111627914,111628190-111628302|11627815-111628302) ---- circRNA异构体的位置长度信息 |
gene_id 基因ID | ensemble id of host gene ---- 基因的ensemble ID |
gene_name 基因名称 | HGNC symbol of host gene ---- 基因的名称 |
gene_type 基因类型 | type of host gene in the annotation gtf file ---- 基因的类型 |
2) 表达矩阵
test_collpase.expression: 包含所有样本中circRNA的表达水平,tsv文件格式。
test_collpase.isoforms:包含所有样本中每个circRNA异构体(isoform)使用指数(index),tsv文件格式。
isoform使用指数公式:
Isoform usage index = Isoform_reads(某个异构体-isoform的数量) / Sum of all isoforms from the same BSJ (共享同一个反向剪切位点的所有异构体-isoform总和)
4. 步骤3:输出文件可视化
从版本v1.1.0以后,CIRI-long包含misc/conver_bed.py 脚本,用户可以使用此脚本将 circRNA.info(gtf格式)转化为.bed格式,此.bed文件可以利用IGV或Jbrowse2软件进行可视化。具体转化代码如下:
$ python3 misc/convert_bed.py collapse_out/sample.info sample_circ.bed四、参考文献
- 专家点评 | 基于纳米孔测序的环形RNA识别和重建新技术
- Chen L-L. The Expanding Regulatory Mechanisms and Cellular Functions of Circular RNAs. Nature Reviews. Molecular Cell Biology, 2020.
- Zheng Y, Ji P, Chen S, et al. Reconstruction of Full-Length Circular RNAs Enables Isoform-Level Quantification. Genome Medicine, 2019, 11(1): 4. Xin R, Gao Y, Gao Y, et al. IsoCirc Catalogs Full-Length Circular RNA Isoforms in Human Transcriptomes. Nature Communications, 2021, 12(1): 266.
- Zhang, J., Hou, L., Zuo, Z., Ji, P., Zhang, X., Xue, Y., & Zhao, F. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nature Biotechnology. (2021).
- CIRI-long 使用文档: https://ciri-cookbook.readthedocs.io/en/latest
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号