VEP — 高效的变异注释工具


工欲善其事必先利其器
1VEP
Ensembl Variant Effect Predictor (VEP) 是由欧洲生物信息研究所(European Bioinformatics Institute, EMBL-EBI)开发的一个高效的基因变异注释工具。VEP是一个强大的工具,其具有以下特性:
- 广泛的注释功能:VEP 可以注释多种类型的变异,包括单核苷酸多态性(SNPs)、插入和删除(indels)、拷贝数变异(CNVs)和结构变异(SVs)。
- 灵活性:用户可以通过命令行界面或网页界面使用VEP,使其适应不同的工作流程和需求。
- 兼容性:VEP 支持多种基因组版本,包括人类、小鼠、斑马鱼等多种物种,便于跨物种比较研究。
- 高效性:VEP 设计用于高效处理大规模数据集,能够快速处理成千上万的变异。
- 定制化输出:用户可以根据需要定制输出格式和内容,例如只选择特定类型的注释或影响。
- 集成其它数据库:VEP 可以集成来自其他数据库的信息,如dbSNP、ClinVar等,为变异提供更全面的生物学背景。
- 插件系统:VEP 提供了一个插件系统,允许用户或开发者添加新的功能或注释来源,以满足特定的研究需求。
官网:https://asia.ensembl.org/info/docs/tools/vep/index.html
文档:http://asia.ensembl.org/info/docs/tools/vep/script/vep_cache.html
github:https://github.com/Ensembl/ensembl-vep
2发表文章
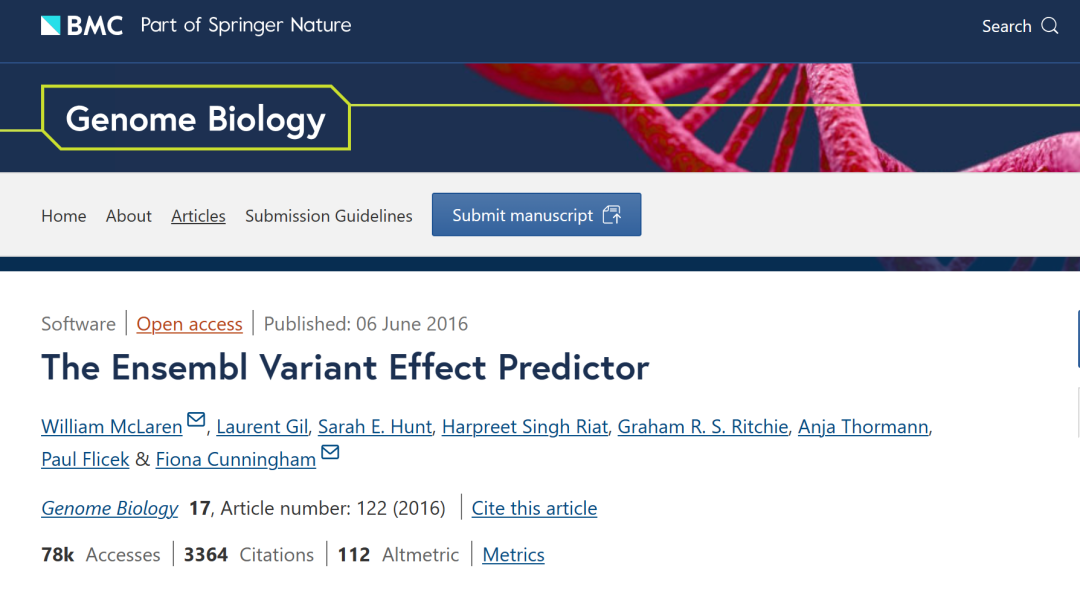
题目: The Ensembl Variant Effect Predictor 期刊:Genome Biology 日期:2016年6月6日 作者&单位:William McLaren & 欧洲生物信息研究所DOI:https://doi.org/10.1186/s13059-016-0974-4
3如何安装
其官网介绍了多种安装方法,我感觉最简单的还是使用Singularity来安装使用。关于Singularity的用法见:Singularity — 生信流程搭建好帮手
mkdir vep
singularity pull --name vep.sif docker://ensemblorg/ensembl-vep
## 下载缓存文件
mkdir $HOME/vep_data
singularity exec vep.sif vep --dir $HOME/vep_data --help
singularity exec vep.sif INSTALL.pl -c $HOME/vep_data -a cf -s homo_sapiens -y GRCh38
singularity exec vep.sif vep --dir $HOME/vep_data -i examples/homo_sapiens_GRCh38.vcf --cache ##示例数据,可不下载
下载成功
当然,除了使用安装程序来下载 INSTALL.pl 也可以使用从其官方链接直接下载:
- https://ftp.ensembl.org/pub/release-111/variation/indexed_vep_cache/
cd $HOME/.vep
curl -O https://ftp.ensembl.org/pub/release-111/variation/indexed_vep_cache/homo_sapiens_vep_111_GRCh38.tar.gz
tar xzf homo_sapiens_vep_111_GRCh38.tar.gz
4功能简述
VEP —— 高效的基因变异注释工具,可以快速地确定变异在基因组中的位置、影响的转录本以及变异对蛋白质功能的可能影响,例如导致蛋白质结构的改变或功能丧失。同时它可以处理多种类型的变异,包括单核苷酸变异(SNVs)、插入删除(indels)、拷贝数变异(CNVs)等。
5基本使用
## 最小化命令
./vep --cache -i input.txt -o output.txt
--cache # 让VEP通过本地缓存来加速注释过程。
#在非脊椎动物运行,需要额外添加一些参数
--genomes # 选项是为了指示VEP连接到正确的数据库服务器,这个服务器专门存储非脊椎物种的基因组数据。这个选项确保VEP能够访问并使用适合给定物种的数据库
##比如对小麦的变异注释(triticum_aestivum)
./vep -i input.txt -o output.txt --species triticum_aestivum --database --genomes
--species # 指定物种
##对于细菌或原生生物 (protists_euglenozoa1)
./vep -i input.txt -o output.txt --species protists_euglenozoa1 --database --genomes --is_multispecies 1
--is_multispecies 1 # 告诉VEP该数据库包含多种物种的基因组数据,其会按照特定方式处理这些数据
## 指定特定版本号
./vep -i input.txt -o output.txt --species triticum_aestivum --cache --cache_version 42
#对于非脊椎物种,Ensembl Genomes的版本号与脊椎动物的版本号不同。--cache_version 选项允许用户指定正确的Ensembl Genomes版本号
singularity 调用示例
singularity exec vep.sif \
vep --dir $HOME/vep_data \
--cache --offline --format vcf --vcf --force_overwrite \
--input_file input/my_input.vcf \
--output_file output/my_output.vcf \
--plugin NMD
--dir #指定vep使用的缓存数据目录
--offline # 启用离线模式,不会建立数据库连接,需要已下载好本地缓存文件
--format vcf:# 指定输入文件的格式为VCF(Variant Call Format
--vcf # 指定输出格式为VCF
--force_overwrite:# 如果输出文件已存在,允许VEP覆盖它
--plugin NMD:# 指定VEP使用NMD(Nonsense-Mediated Decay)插件。NMD插件用于预测变异是否会触发无义介导的mRNA降解机制
若不使用官方提供的预注释文件
官方预处理注释文件下载:https://ftp.ensembl.org/pub/release-111/variation/indexed_vep_cache/
对于VEP没有预先提供注释文件的物种或者是新的、特定的基因组组装版本、用户需要在注释过程中使用经过定制的基因组特征或注释等情况下,用户可以自己提供GFF或GTF文件以及相应的基因组序列(FASTA文件)。不过GTF或GFF文件必须按染色体顺序排序,删除header信息且文件需要进行bgzip压缩并用tabix进行索引
##GFF文件
grep -v "#" data.gff | sort -k1,1 -k4,4n -k5,5n -t$'\t' | bgzip -c > data.gff.gz
tabix -p gff data.gff.gz
./vep -i input.vcf --gff data.gff.gz --fasta genome.fa.gz
## 与缓存注释文件一起使用(多个注释文件)
./vep -i input.vcf --cache --gff data.gff.gz --fasta genome.fa.gz
##GTF件同上
./vep -i input.vcf --gtf data.gtf.gz --fasta genome.fa.gz
./vep -i input.vcf --cache --gtf data.gtf.gz --fasta genome.fa.gz
自定义注释输出
使用逗号分隔来配置每个自定义文件 键值对列表
singularity exec vep.sif \
vep --dir $HOME/vep_data \
--cache --offline --format vcf --vcf --force_overwrite \
--input_file input/my_input.vcf \
--output_file output/my_output.vcf \
--custom file=/path/to/custom_files/clinvar.vcf.gz,short_name=ClinVar,format=vcf,type=exact,coords=0,fields=CLNSIG%CLNREVSTAT%CLNDN
--custom # 允许用户添加自定义注释
更多用法详见:https://useast.ensembl.org/info/docs/tools/vep/script/vep_options.html
支持web界面
Web界面:https://asia.ensembl.org/info/docs/tools/vep/online/index.html
感兴趣的用户可以去摸索一下
提交表单
参考:
- http://asia.ensembl.org/info/docs/tools/vep/script/vep_tutorial.html
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-04-02,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号