BD Rhapsody上游定量流程


工欲善其事必先利其器
单细胞技术的核心是能够从单一细胞获取高通量的基因表达数据。与传统的基因表达分析相比,它不是测量一个样本中成千上万细胞的平均表达,而是能够揭示个别细胞之间的差异,这对于理解组织中的微环境、细胞类型的多样性及其功能至关重要。而对于单细胞转录组学技术,除了大火的10X单细胞技术以外,另一个就是由 Becton, Dickinson and Company开发的BD Rhapsody 。BD Rhapsody 系统通过以下几个主要步骤来实现单细胞转录组分析:
- 细胞捕获和制备:首先,单个细胞被捕获并封装到微流控芯片的微孔中。每个微孔还包含了用于逆转录的全部所需试剂,允许在单细胞水平上进行mRNA的捕获和转录成cDNA。
- 标记和扩增:利用BD Rhapsody 的微球技术,每个微球都被标记有唯一的分子标记(UMI)和细胞标记,这使得在后续的步骤中可以追踪每个cDNA分子来源于哪个细胞。随后进行cDNA的扩增。
- 测序准备:扩增后的cDNA被制备成适合测序的文库,通过下一代测序(NGS)平台进行高通量测序。
- 数据分析:最后,生成的序列数据被用于定量分析每个细胞的基因表达水平,可以利用各种生物信息学工具和算法来识别细胞类型、细胞状态、细胞间相互作用以及基因表达的调控网络等。
那么今天就来学习一下 BD Rhapsody 的上游定量流程
1首先是准备分析环境
安装 cwltool
mamba create -n BD python=3.11.3
mamba activate BD
pip install cwlref-runner
##如果pip安装失败,可以选择国内镜像安装
pip install cwlref-runner -i https://pypi.douban.com/simple/
下载 BD官方pipeline 文件
BD Genomics Rhapsody Analysis pipeline 网址:https://bitbucket.org/CRSwDev/cwl/src/master/v2.0/
v2.0
需要注意的一点是运行 rhapsody_pipeline_2.0cwl 这个是需要服务器上有配置好Docker且你有Docker权限的。可以简单看一下代码,是有用到docker的

rhapsody_pipeline_2.0cwl
2其次是下载参考基因组文件
网址:http://bd-rhapsody-public.s3-website-us-east-1.amazonaws.com/Rhapsody-WTA/Pipeline-version2.x_WTA_references/

参考基因组文件
##humam
wget -c https://bd-rhapsody-public.s3.amazonaws.com/Rhapsody-WTA/Pipeline-version2.x_WTA_references/RhapRef_Human_WTA_2023-02.tar.gz
##mouse
wget -c https://bd-rhapsody-public.s3.amazonaws.com/Rhapsody-WTA/Pipeline-version2.x_WTA_references/RhapRef_Mouse_WTA_2023-02.tar.gz
3最小化使用
yml文件修改
BD Rhapsody 上游定量流程其实已经封装的很好,环境及所需文件准备好后只需简单修改pipeline_inputs_template_2.0.yml 即可
主要修改Reads 【指定输入文件】和 Reference_Archive 【指定参考基因组文件】这个两个部分(如图所示)

pipeline_inputs_template_2.0.yml
更多pipeline的可选参数的修改见:https://bd-rhapsody-bioinfo-docs.genomics.bd.com/setup/input/parameters.html
提交运行
对于单个样本
yml文件的输入文件格式要求:
## 单个样本
Reads:
- class: File
location: "/home/project/b_rawdata/download/JZsc603/JZsc603_S33_L002_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc603/JZsc603_S33_L002_R2_001.fastq.gz"
## 单个样本多个文件
Reads:
- class: File
location: "/home/project/b_rawdata/download/JZsc501/JZsc501_S13_L001_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc501/JZsc501_S39_L004_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc501/JZsc501_S13_L001_R2_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc501/JZsc501_S39_L004_R2_001.fastq.gz"
运行脚本run_BD.sh 写入以下内容
#! /bin/bash -xe
/home/username/miniconda3/envs/BD/bin/cwl-runner \
--outdir /home/project/test603 \
--tmpdir-prefix /home/project/sc603 \
/home/script/rhapsody_pipeline_2.0.cwl \
/home/script/test603.yml #pipeline_inputs_template_2.0.yml修改后的文件
修改完 yml 文件提交后台运行即可
mmaba activate BD
nohup bash run_BD.sh 1>test501.log 2>&1 &
如果是多个样本
多样本的话,需要注意一下reads文件在yml文件的写法。
如果按以下方法给定输入文件:
Reads:
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R1_001.fastq.gz"
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R1_001.fastq.gz"
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R2_001.fastq.gz"
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R2_001.fastq.gz"
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R2_001.fastq.gz"
会报错 found duplicate key "location" 。
正确的写法应该是:
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R2_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R2_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R2_001.fastq.gz"
或者
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R1_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1641/JZsc1641_S3_L003_R2_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc1672/JZsc1672_S3_L004_R2_001.fastq.gz"
- class: File
location: "/home/project/b_rawdata/download/JZsc875/JZsc875_S18_L002_R2_001.fastq.gz"
多样本同时运行也仅需修改yml文件的输入即可,提交运行的命令同上
4结果文件
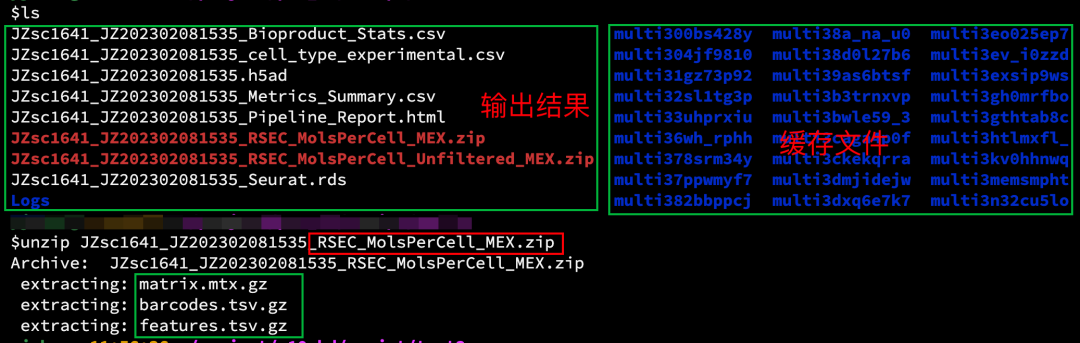
输出
通常结果包含以下文件(不同参数,会有些许出入)
[sample_name]_Metrics_Summary.csv : 测序、分子和细胞指标的报告,包含每个样品的测序质量、检测到的总分子数和细胞数等信息。
[sample_name]_Pipeline_Report.html :测序分析运行结果的摘要报告
[sample_name]_RSEC_MolsPerCell_MEX.zip 和 [sample_name]_DBEC_MolsPerCell_MEX.zip: 基于RSEC或DBEC计算的每个细胞的生物产品(bioproduct)分子数量的数据表
[sample_name]_RSEC_MolsPerCell_Unfiltered_MEX.zip: 包含未经过滤的数据表,这些数据表列出了所有细胞标签以及有至少10次读数的细胞信息
[sample_name].BAM 和 [sample_name].BAM.bai: R2 reads的比对信息,默认参数为节省空间是不输出。
[sample_name]_Seurat.rds :RSEC分子数据表和所有细胞注释元数据的Seurat(.rds)格式文件,用于R的Seurat包进行下游分析
[sample_name].h5mu或 [sample_name].h5ad: RSEC分子数据表和所有细胞注释元数据的Scanpy(.h5ad)/Muon(.h5mu)格式文件,用于Python的Scanpy包或其它兼容工具进行下游分析
[sample_name]_Bioproduct_Stats.csv :基于RSEC和DBEC唯一分子标识符(UMI)调整算法的生物产品统计数据
注:(a) 如果是多重样本运行,数据表中包含了所有样本合并后的推测细胞数。(b) DBEC数据表只有在包括针对mRNA或AbSeq生物产品目标的实验时才有输出。
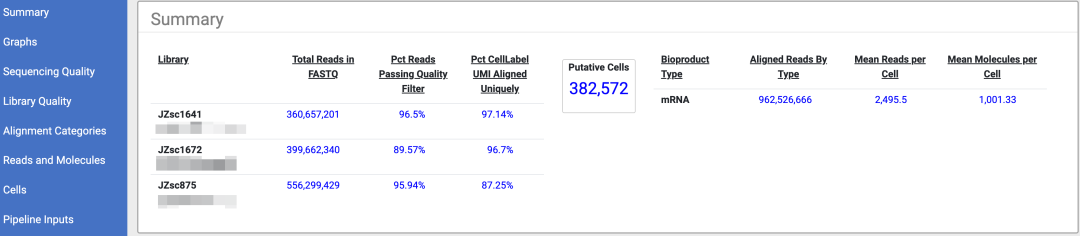
报告摘要-部分
通常来说,下游分析我们可以使用[sample_name]_Seurat.rds 这个文件走Seurat分析流程 ,但是由于目前rhapsody_pipeline_2.0cwl 这个pipeline封装的还是Seurat V4版本 ,所以该输出结果是Seurat V4的数据格式 。
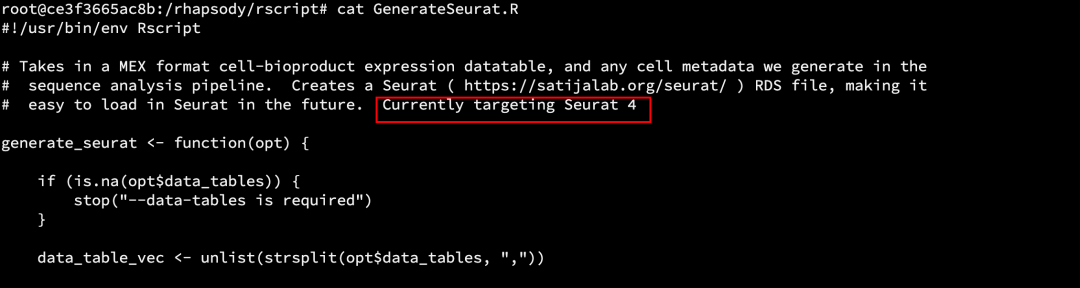
docker内的调用脚本
如果你想直接使用 SeuratV5 进行下游分析,其实也只需解压 [sample_name]_RSEC_MolsPerCell_MEX.zip 这个文件,正常读入文件即可进行后续分析。
关于MEX格式
MEX是单细胞技术中一种稀疏矩阵的表示方法。单细胞数据处理后通常以两种方式存储:HDF5格式或者MEX格式。
MEX是Market Exchange Format的缩写,通常包含以下几个文件来有效地存储和描述矩阵数据:
- 表达矩阵文件(matrix.mtx.gz):这是最重要的文件,包含了基因表达数据。在稀疏格式下,它仅存储非零表达值及其在矩阵中的位置(即,哪个基因在哪个细胞中表达)。文件格式是
.mtx,代表Matrix Market格式,这是一种广泛支持的标准稀疏矩阵格式。 - 基因标识符文件(features.tsv.gz):通常是一个文本文件,列出了表达矩阵中每一行对应的基因。这个文件允许将表达矩阵中的行映射到实际的基因名称或ID。
- 样本或细胞标识符文件(barcodes.tsv.gz):另一个文本文件,列出了表达矩阵中每一列对应的细胞。这使得研究者可以知道每一列数据对应的具体细胞样本。
MEX文件的特点是:
- 允许以百分号开头的注释行;
- 对于稀疏矩阵,使用“坐标”格式;
- 对于一般的密集矩阵,使用“数组”格式。
使用这种稀疏矩阵的格式的优势:
- 空间效率:因为大多数基因在大多数细胞中的表达量为零,稀疏矩阵格式允许仅存储非零数据点,大大减少了所需的存储空间。
- 计算效率:与完整矩阵相比,处理稀疏矩阵的算法可以显著提高,因为它们可以跳过大量的零值计算。
- 可扩展性:这种格式使得处理大规模数据集更加可行,特别是随着单细胞测序技术的快速发展和应用。
参考:
- https://bd-rhapsody-bioinfo-docs.genomics.bd.com/
- https://mp.weixin.qq.com/s/E4K0qOTmftajLcdEZ9FPyw
- https://mp.weixin.qq.com/s/Mdpdu1bGfgNYzPO0oi3lDQ
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-04-09,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号