好狠的心啊,直接就删除8成的单细胞!



Following quality control analyses, filter- ing and doublet removal, 14,504 cells remained for characterization.
数据集是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232309
可以看到是:
GSM7325156 Ovary, 3 months, 1
GSM7325157 Ovary, 3 months, 2
GSM7325158 Ovary, 3 months, 3
GSM7325159 Ovary, 3 months, 4
GSM7325160 Ovary, 9 months, 5
GSM7325161 Ovary, 9 months, 6
GSM7325162 Ovary, 9 months, 7
GSM7325163 Ovary, 9 months, 8
很清晰的文件结构:
很容易整理它们后读取,常规的单细胞转录组降维聚类分群代码可以看 :链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程。
dir='GSE232309_RAW/outputs/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
print(dim(ct))
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
可以看到的是原始的8个10x单细胞样品的barcodes数量是:
[1] "GSM7325156_1"
[1] 32285 775390
[1] "GSM7325157_2"
[1] 32285 941662
[1] "GSM7325158_3"
[1] 32285 1162144
[1] "GSM7325159_4"
[1] 32285 1139625
[1] "GSM7325160_5"
[1] 32285 959979
[1] "GSM7325161_6"
[1] 32285 868379
[1] "GSM7325162_7"
[1] 32285 915225
[1] "GSM7325163_8"
[1] 32285 911686
粗浅的过滤后是67268个细胞,如下所示:
> do.call(rbind,lapply(sceList, dim))
[,1] [,2]
[1,] 19408 6630
[2,] 20115 8270
[3,] 21212 24281
[4,] 21458 14026
[5,] 20568 2981
[6,] 19651 4062
[7,] 20468 3778
[8,] 19953 3240
如果我们就使用上面的粗浅的过滤结果,常规的质量控制过滤后做降维聚类分群:
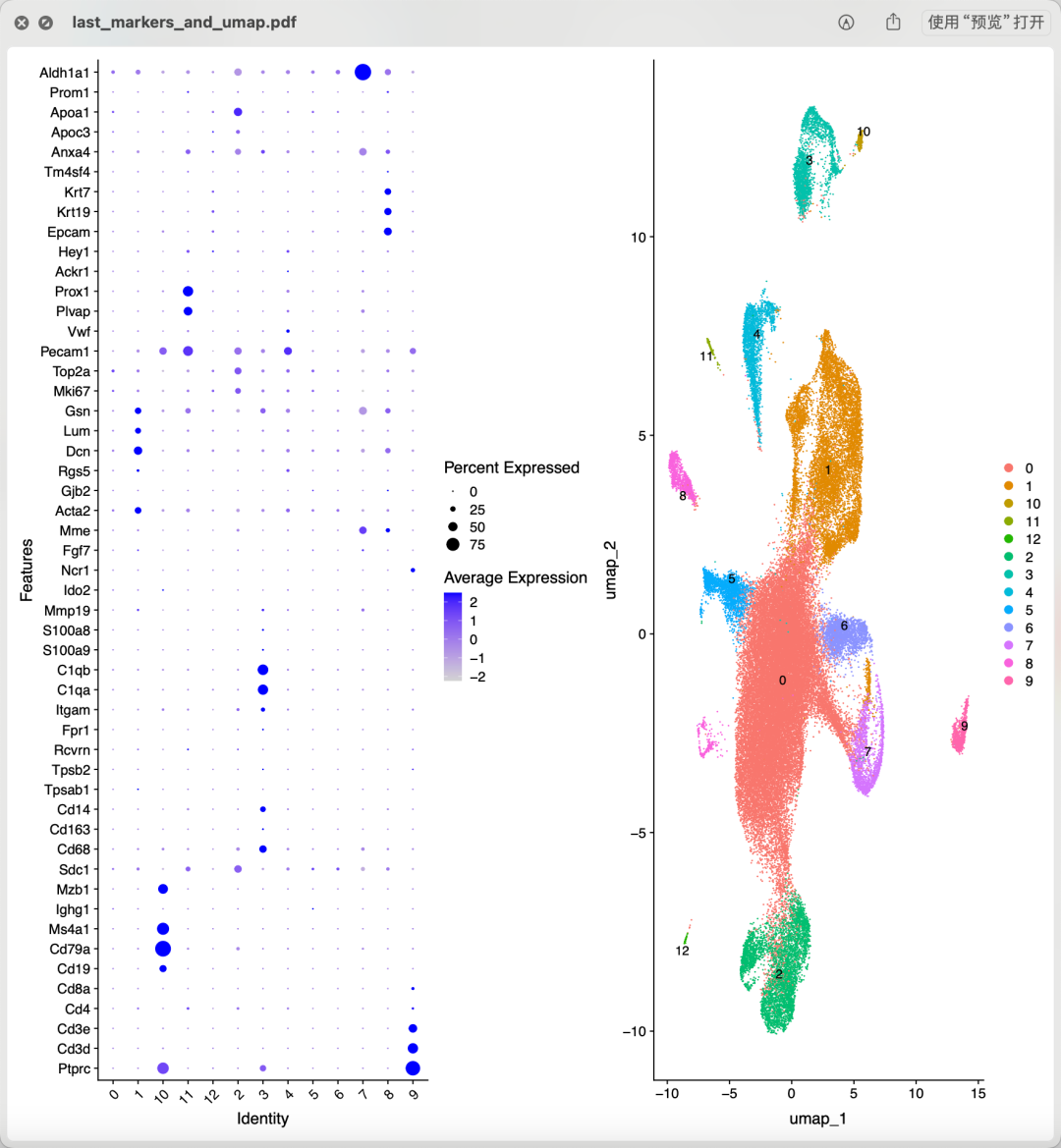
常规的质量控制过滤
从上面混成一团的降维聚类分群结果看应该是各个样品的各个细胞的质量普遍不行,所以需要进行比较严格的质量控制和过滤。当然了,使用什么样的阈值就仁者见仁智者见智啦。如果想达到文章提到的 14,504 cells remained for characterization,就相当于是直接就删除8成的单细胞。我也测试了几个过滤指标,最后也是同样的细胞数量,如下所示降维聚类分群:
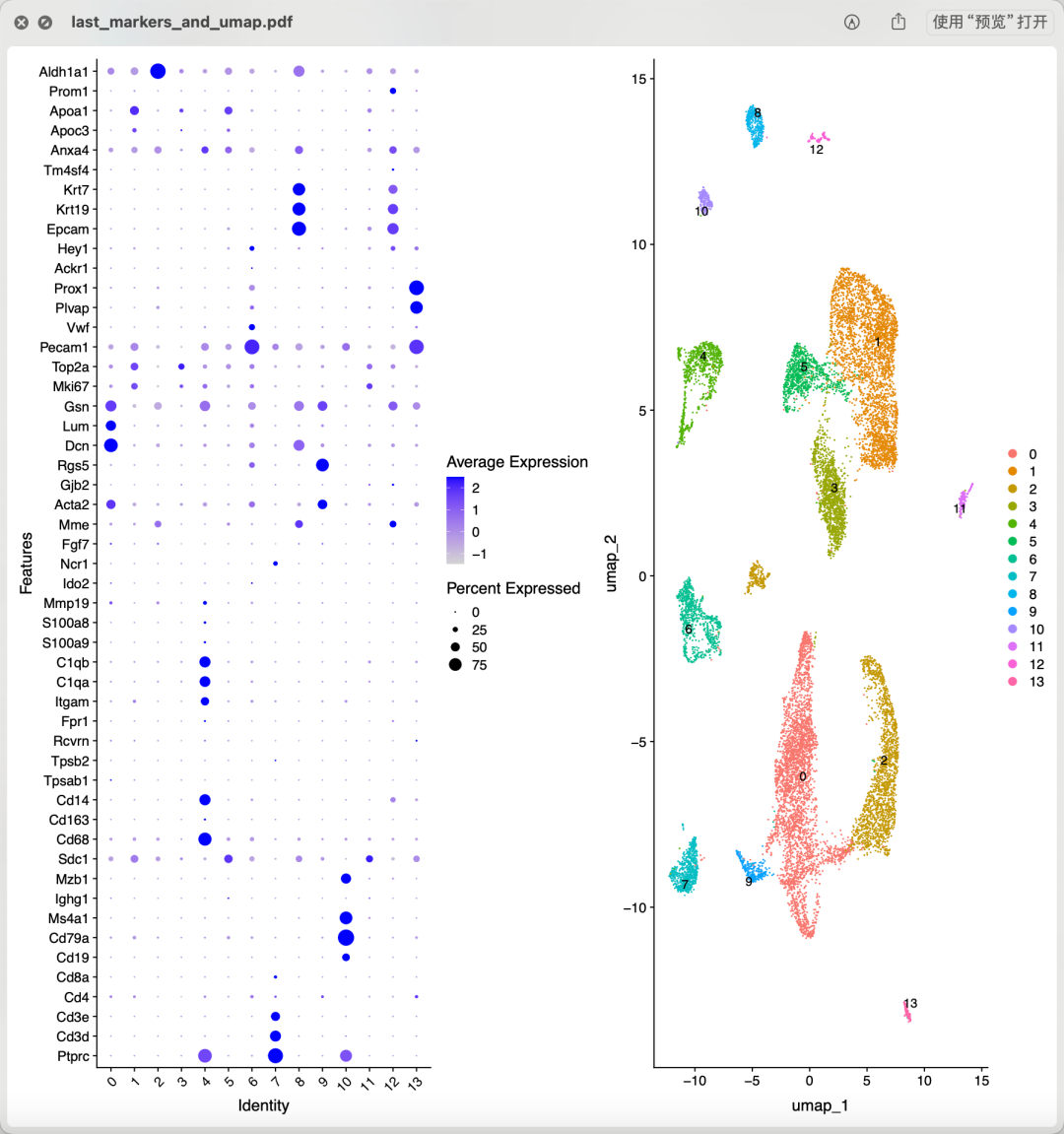
同样的细胞数量
跟文章里面的分群结果不太一样:
- stromal cells (n = 5,671), which segregated into three CLUs.
- Stroma A, was characterized by having a major Col1a1
- Stroma B, was identified by the expression of several stromal markers (Bgn, Ogn, Dcn, Lum, Col1a1
- Stroma C, was characterized by Notch3
- The second most common cell type was found to be GCs (n = 3,334),
- theca cells (TCs; n = 1,637; Srd5a1
- phagocytes (two distinct CLUs; n = 1,099; C1qa
- endothelial cells (n = 798; Cd34
- T lymphocytes (n = 728; Cd3g
- epithelial cells (two distinct CLUs; n = 450; Upk1b or Gpm6a
- oocytes (n = 224; Zp3
- luteal cells (n = 206; Ptgfr )
- B lymphocytes (n = 202; Cd79a
好玩的是文章把单细胞亚群取名为 these clusters (CLUs)
如果是你遇到了这么惨的数据
首先你有勇气删除这么多吗,其次你还有勇气做下去吗?
这个2024的文章:《A single-cell atlas of the aging mouse ovary》,不仅仅是“硬着头皮”进行了降维聚类分群,而且还在进行第二层次的降维聚类分群(免疫细胞细分):
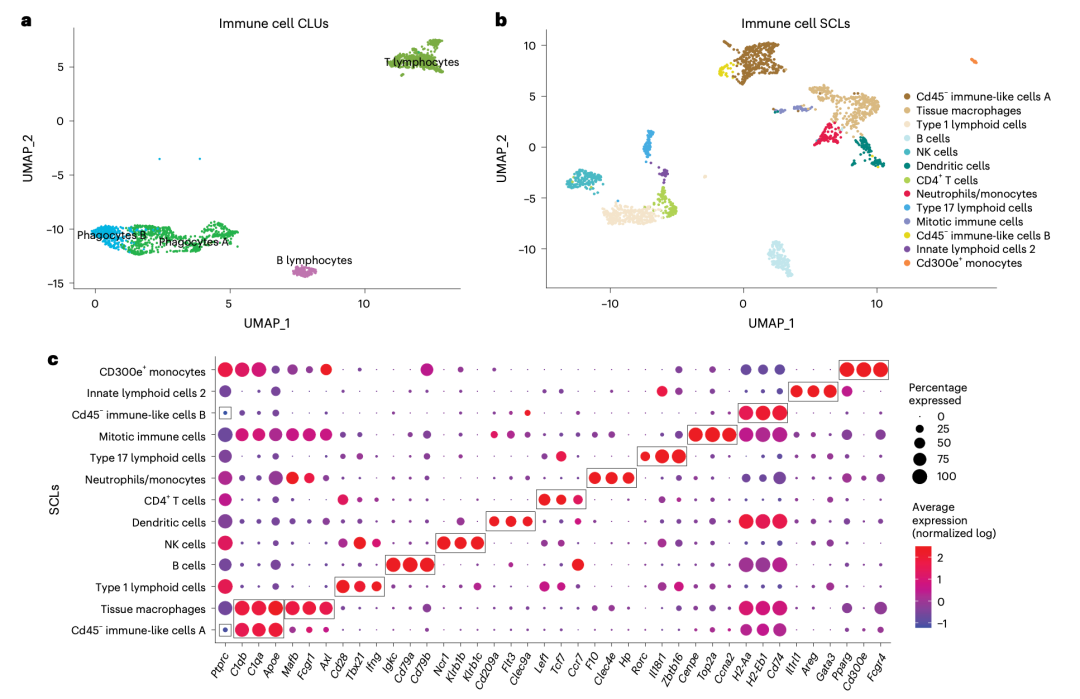
降维聚类分群(免疫细胞细分)
而且看起来还蛮清晰的哦,细分之后就可以继续看两个分组的各个细分亚群比例差异找到写作的点啦!
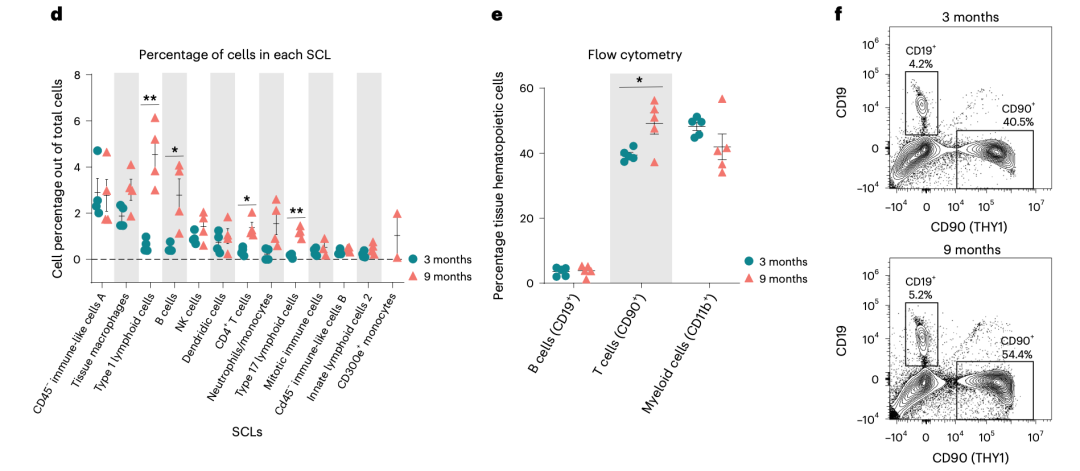
两个分组的各个细分亚群比例差异
相当于是某种程度的变废为宝吧!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-05-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号