SAHMI 单细胞宿主-微生物互作分析代码实战


编者按:同为显微镜献给现代生物学的礼物,对单细胞和微生物的观察一直相伴相生,它们一起定义着免疫、互作以及生命的基本单元。随着测序技术的到来,一个是宏基因组测序和扩增子测序打开微生物组的世界,一个是单细胞测序打开的细胞图谱画卷,堪称单细胞与微生物的“第二次亲密接触”,这一次的火花将更加热烈。
2020年11月29日,拙文《单细胞时代 || 宿主-微生物组相互作用》中,浅谈了在单细胞水平分析宿主细胞与微生物组的相互作用,当时主要参考的文章是:Host-Microbiome Interactions in the Era of Single-Cell Biology。
近四年来,在这个领域又有许多喜人的进展:
- 实验技术方面:开发出可以同时对宿主单细胞及其微生物测序的新技术。如Angewandte Chemie2024年5月发表的“High-Throughput Host-Microbe Single-Cell RNA Sequencing Reveals Ferroptosis-Associated Heterogeneity during Acinetobacter baumannii Infection”文章。
- 生信技术方面:开发出基于单细胞转录组数据或联合宏基因组数据分析的新方法,如Nature Computational Science发表的“Denoising sparse microbial signals from single-cell sequencing of mammalian host tissues”方法。
- 当然,这些离不开肿瘤内微生物越来越被重视的事实。将来我们再谈肿瘤微环境,如果没有提到其中的微生物组,那是将不完整的。如2024年4月,Cell上发表的“A pan-cancer analysis of the microbiome in metastatic cancer”,作者通过多组学手段揭示转移性肿瘤队列的泛癌微生物组。
2024年5月底,《国家自然科学基金医学科学“十四五”学科发展战略研究报告》指出:我国未来医学需重点布局的领域方向主要有九个方面,其中前三个是:
- 一是疾病图谱的绘制:利用高精度单细胞多组学测序技术,系统解析疾病发生发展过程中基因表达调控网络和表观遗传调控网络的动态变化过程。
- 二是疾病免疫异常机制及免疫治疗策略的研究:研究自身免疫性疾病发生机制、免疫异常机理;研发治疗新策略,解析肿瘤中免疫微环境的构成,逆转肿瘤免疫抑制性微环境。
- 三是微生物组学与人体健康和疾病的关系:识别人体个体化微生物组学特征,阐明肠道微生物影响疾病发生机制及治疗意义。
可见单细胞、免疫、微生物是基础性比较强的技术领域,如何在这些技术之间找到结合点或融合的地方?是值得我们思考。在技术发展路径上,解决方案这个维度上,单细胞、免疫、微生物都是当下生命科学比较重要的发展前沿。
鉴于单细胞测序和微生物测序在生物信息分析方面有着惊人的相似性,微生物本身是单细胞,而单细胞测序也可以分析微生物信息,比如今天我们要踩坑的SAHMI(Single-cell Analysis of Host-Microbiome Interactions) 方法。
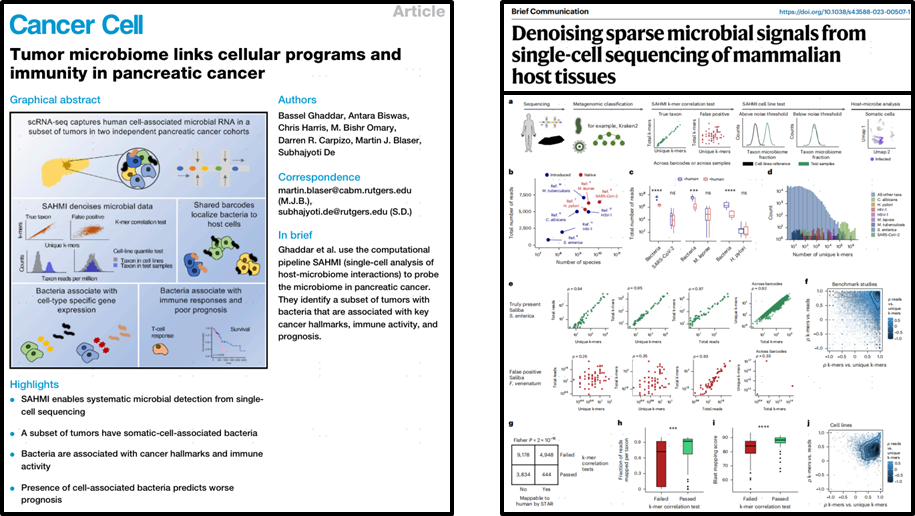
仅从单细胞转录组数据,不需要单独做额外的测序,就可以识别细胞亚群特异的胞内微生物丰度情况,得到的经典结果是一张映射微生物丰富的UMAP图,还可以在这个基础上进行其他分析。
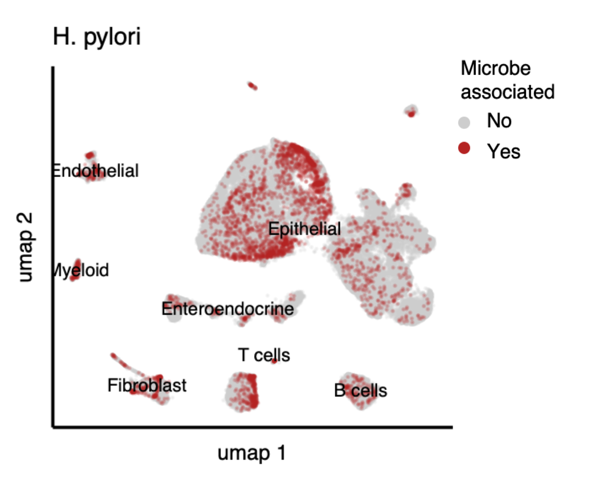
这也是异质性的一种扩展。卖个关子,留一个开放性问题,就像用单细胞转录组一样,在单细胞水平上分析胞内菌是不是可以称作:单细胞微生物组?分辨率加一种组学的构词法。
好了,切入正题,SAHMI是一个计算框架,用于从单细胞数据中识别真正存在的微生物序列,可以正确识别存在于不同组织中的已知微生物感染。应用于单细胞和空间基因组数据,从而实现体细胞和微生物的联合分析。
我们打开原文,Denoising sparse microbial signals from single-cell sequencing of mammalian host tissues,找到代码。在这篇文章中,我们主要是根据作者提供的代码,找到每个barcode对应的微生物丰度,将其映射在单细胞的UMAP上。
这里假设,读者诸君已经了解git、R语言、Seurat、以及微生物组和单细胞测序数据结构的基本知识。
git clone https://github.com/sjdlabgroup/SAHMI.git
代码下载完之后,我们看看这里都有哪些文件。
$ tree -L 2
.
├── exampledata
│ ├── SRR9713132.kraken.report.txt
│ ├── SRR9713132.sckmer.txt
│ └── zhang.reports.RDS
├── Fig. 1A.png
├── Fig. 2.png
├── functions
│ ├── extract_microbiome_output.r
│ ├── extract_microbiome_reads.r
│ ├── kreport2mpa.py
│ ├── mytest
│ ├── mytest.sh
│ ├── mytest.V
│ ├── read_kraken_reports.r
│ ├── run_kraken.r
│ ├── sckmer.r
│ ├── sckmer_unpaired.r
│ └── taxa_counts.r
├── LICENSE.MD
├── PDACdata
│ ├── pdac_sahmi.RDS
│ ├── peng_microbiome.RDS
│ ├── README.md
│ ├── rutgers_sahmi.RDS
│ ├── steele_microbiome.RDS
│ └── Table S3.xlsx
├── README_files
│ └── figure-markdown_github
├── README.md
├── README.Rmd
└── TableS4.xlsx
可以看出SAHMI基本上是由几个R/Python的脚本组成的并不是一个完整的R包或python的库。这就需要我们把对应的R包和版本安装好,最好是为这个分析创建一个全新的conda环境。
我们根据github上的教程知道了这些脚本主要执行的功能,一个主要的脚本是:run_kraken.r。我们运行帮助文档,看看R包有没有配置齐。
Rscript functions/run_kraken.r --help
Usage: functions/run_kraken.r [options]
Options:
--sample=SAMPLE
sample name
--fq1=FQ1
path to fastq 1 file
--fq2=FQ2
path to fastq 2 file
--out_path=OUT_PATH
output directory path
--ncbi_blast_path=NCBI_BLAST_PATH
path to ncbi-blast
--Kraken2Uniq_path=KRAKEN2UNIQ_PATH
path to Kraken2 main 'kraken2' function
--kraken_database_path=KRAKEN_DATABASE_PATH
path to kraken database
--kreport2mpa_path=KREPORT2MPA_PATH
path to kreport2mpa.py' function
--paired=PAIRED
paired-end fastq files (T) or sinle-end (F)
-h, --help
Show this help message and exit
可以看到,在我的环境中是可以运行帮助信息的,这里建议大家打开这些脚本,看看哪些R包比较陌生以及输入参数的基本处理。这些步骤不能跳过,不能代劳,不可忽视。
从这个帮助文档中,我们看到依赖的一个熟悉的软件:Kraken2,这就是在微生物领域中用于宏基因组样本reads的分类注释和物种丰度估计的工作流程。它的GitHub仓库:https://github.com/DerrickWood/kraken2
理论文章如下:
Lu, J., Rincon, N., Wood, D.E. et al. Metagenome analysis using the Kraken software suite. Nat Protoc 17, 2815–2839 (2022). https://doi.org/10.1038/s41596-022-00738-y
下面的主要工作就是安装这个软件,以及kraken2所需要的细菌或真菌等的比对索引构建。安装这部分
git clone https://github.com/DerrickWood/kraken2.git
sh install_kraken2.sh $KRAKEN2_DIR
一切顺利的话,我们的kraken2就安装好了。我们依然使用帮助文档的打印来初步检查安装是否成功。
./kraken2 -h
Usage: kraken2 [options] <filename(s)>
Options:
--db NAME Name for Kraken 2 DB
(default: none)
--threads NUM Number of threads (default: 1)
--quick Quick operation (use first hit or hits)
--unclassified-out FILENAME
Print unclassified sequences to filename
--classified-out FILENAME
Print classified sequences to filename
--output FILENAME Print output to filename (default: stdout); "-" will
suppress normal output
--confidence FLOAT Confidence score threshold (default: 0.0); must be
in [0, 1].
--minimum-base-quality NUM
Minimum base quality used in classification (def: 0,
only effective with FASTQ input).
--report FILENAME Print a report with aggregrate counts/clade to file
--use-mpa-style With --report, format report output like Kraken 1's
kraken-mpa-report
--report-zero-counts With --report, report counts for ALL taxa, even if
counts are zero
--report-minimizer-data With --report, report minimizer and distinct minimizer
count information in addition to normal Kraken report
--memory-mapping Avoids loading database into RAM
--paired The filenames provided have paired-end reads
--use-names Print scientific names instead of just taxids
--gzip-compressed Input files are compressed with gzip
--bzip2-compressed Input files are compressed with bzip2
--minimum-hit-groups NUM
Minimum number of hit groups (overlapping k-mers
sharing the same minimizer) needed to make a call
(default: 2)
--help Print this message
--version Print version information
If none of the *-compressed flags are specified, and the filename provided
is a regular file, automatic format detection is attempted.
直到这里都是好消息,在构建kraken2比对索引的时候,我遇到了内存和计算资源的挑战。根据kraken2文档(https://github.com/DerrickWood/kraken2/wiki/Manual)的描述,我们需要用kraken2-build来完成。
kraken2-build --standard --db $DBNAME
(Replace "$DBNAME" above with your preferred database name/location. Please note that the database will use approximately **100 GB **of disk space during creation, with the majority of that being reference sequences or taxonomy mapping information that can be removed after the build.)
而且速度非常的慢,登陆两天没有什么结果之后,一个偶然的机会,看到可以直接下载别人构建好的索引文件,这个宝藏地址在这里:https://benlangmead.github.io/aws-indexes/k2
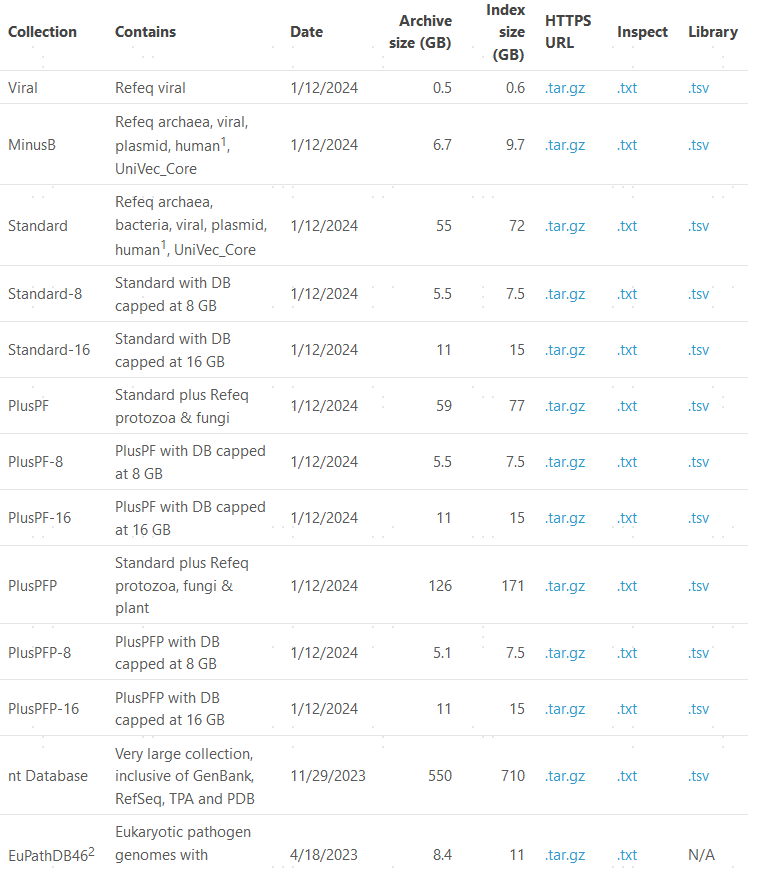
我们综合考虑内存大小和自己想研究的微生物种类,选择了16S_SILVA132_k2db,当然这里的自由是给到大家的,您可以根据自己的情况选择喜欢的索引。下载了任何一个东西,我的习惯都是要看看自己下载的是什么,有没有可读的文件。
16S_SILVA132_k2db/
├── database100mers.kmer_distrib
├── database150mers.kmer_distrib
├── database200mers.kmer_distrib
├── database250mers.kmer_distrib
├── database50mers.kmer_distrib
├── database75mers.kmer_distrib
├── hash.k2d
├── opts.k2d
├── README.md
├── seqid2taxid.map
└── taxo.k2d
这里可以读的就是README文件,告诉我们这个索引可以如何用。
好了,到这里我们为运行SAHMI的核心部件配置齐了。下面还差测试数据。这里我们拿一个单细胞转录组数据,它由R1和R2组成。
下面我们开始测试SAHMI的各个部分。第一部分是微生物的分类注释
单细胞转录组测序的fastq文件可以用于宏基因组分类。这一步骤使用基于k-mer的映射进行,该映射可以识别每个k-mer和read的微生物注释ID。SAHMI被优化为与Kraken2Uniq一起运行,Kraken2Uniq可以在参考宏基因组数据库中找到候选35-mer基因组子串与基因组最低共同祖先的精确匹配。在这一阶段,必须将所有实际可能的基因组(例如宿主、已知载体等)作为映射参考,或者排除宿主可映射的reads。
mkdir mytest/
Rscript run_kraken.r \
--sample mytest \
--fq1 /home/data/software/tinygex_S1_L001_R1_001.fastq.gz \
--fq2 /home/data/software/tinygex_S1_L001_R2_001.fastq.gz \
--out_path mytest/ \
--Kraken2Uniq_path /home/data/software/kraken2/kraken2 \
--kraken_database_path /home/data/software/kraken2/Kraken2DB/16S_SILVA132_k2db \
--kreport2mpa_path /home/data/software/SAHMI/functions/kreport2mpa.py \
--paired T
我们的数据比较小,运行很快。
Loading database information... done.
230610 sequences (27.44 Mbp) processed in 1.332s (10390.9 Kseq/m, 1236.52 Mbp/m).
17051 sequences classified (7.39%)
213559 sequences unclassified (92.61%)
得到的结果如下:
$ tree mytest
mytest
├── mytest_1.fq
├── mytest_2.fq
├── mytest.kraken.output.txt
├── mytest.kraken.report.mpa.txt
├── mytest.kraken.report.std.txt
└── mytest.kraken.report.txt
一定要看看跑出来的结果是怎么样的,我就不再一一cat了。
接下来使用以下脚本从fastq文件和Kraken输出文件中提取Microbiome读数:
Rscript extract_microbiome_reads.r \
--sample_name mytest_1 \
--fq mytest/mytest_1.fq \
--kraken_report mytest/mytest.kraken.report.txt \
--mpa_report mytest/mytest.kraken.report.mpa.txt \
--out_path mytest/
Rscript extract_microbiome_reads.r \
--sample_name mytest_2 \
--fq mytest/mytest_2.fq \
--kraken_report mytest/mytest.kraken.report.txt \
--mpa_report mytest/mytest.kraken.report.mpa.txt \
--out_path mytest/
其实这里我有些困惑,R1里面都是barcode和UMI也没有微生物的序列,所以为什么也需要跑这个?多生成了两个序列文件。
mytest_1.fa
mytest_2.fa
$ head mytest_2.fa
>A00228:279:HFWFVDMXX:1:1356:17173:26443 3:N:0:0 kraken:taxid|3303
GTTCTCTATGCAACTTCCAATAAAACCTCTTCATTTGAAAGGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
>A00228:279:HFWFVDMXX:1:2462:28980:21402 3:N:0:0 kraken:taxid|2805
GAATGGAATGGAATTGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATTGAATGGAATGGAATGGAATGGAATGGAAAGGAATGT
>A00228:279:HFWFVDMXX:1:2175:8648:34851 3:N:0:0 kraken:taxid|28455
GGTCGCTCGCTCCTCTCCTACTTGGATAACTGTGGTATTTCTAGAGCTAATACATGCCGACGGGCGCTGACCCCCTTCGCGGGGGGGATGC
>A00228:279:HFWFVDMXX:1:1366:11189:33019 3:N:0:0 kraken:taxid|7824
TCCAGCACCAATAGCTTATATTAAAGTTGCTGCAGTTAAAAAGCTCGTAGTTGGAAATTCGTAGCTGGCGGTCGGTCCGCAGCGAGGCTAG
>A00228:279:HFWFVDMXX:1:1459:4734:18270 3:N:0:0 kraken:taxid|7708
GCGCCCCCTCGATTCTCTTAGCTGAGTTTCCCTCGGGGCCCGAATAGTTTACTTTGAAAAAATTAGATTGTTCAAAGCAGGCCCGATACGC
可以看到我们的序列都加上了微生物的taxid信息。
Rscript extract_microbiome_output.r --sample_name mytest --output_file mytest/mytest.kraken.output.txt --kraken_report mytest/mytest.kraken.report.txt --mpa_report mytest/mytest.kraken.report.mpa.txt --out_path mytest/
$ head mytest/mytest.microbiome.output.txt
C A00228:279:HFWFVDMXX:1:1356:17173:26443 Gammaproteobacteria (taxid 3303) 28|91 |:| 0:37 3:5 3303:15
C A00228:279:HFWFVDMXX:1:2462:28980:21402 Mitochondria (taxid 2805) 28|91 |:| 0:12 2805:8 0:7 2805:5 0:22 2805:3
C A00228:279:HFWFVDMXX:1:2175:8648:34851 Malassezia (taxid 28455) 28|91 |:| 4:6 0:6 4:1 0:1 4:3 0:1 28455:4 4:35
C A00228:279:HFWFVDMXX:1:1366:11189:33019 Taphrina (taxid 7824) 28|91 |:| 0:2 7824:3 0:11 4:10 0:1 9379:2 0:28
C A00228:279:HFWFVDMXX:1:1459:4734:18270 Saccharomycetales (taxid 7708) 28|91 |:| 0:29 13174:1 0:10 12972:3 7708:6 0:8
C A00228:279:HFWFVDMXX:1:2266:22155:7044 Basidiomycota (taxid 7825) 28|91 |:| 4:26 1:15 0:3 7825:2 0:11
C A00228:279:HFWFVDMXX:1:2465:30915:1924 Basidiomycota (taxid 7825) 28|91 |:| 0:4 4:16 1:15 0:3 7825:2 0:17
C A00228:279:HFWFVDMXX:1:2337:5122:8140 Chrysomyxa (taxid 8427) 28|91 |:| 9444:2 8427:2 4:6 6428:7 13043:1 1:15 4:3 7825:2 0:19
C A00228:279:HFWFVDMXX:1:2331:27742:33411 Basidiomycota (taxid 7825) 28|91 |:| 4:19 0:10 4:3 7825:2 0:23
C A00228:279:HFWFVDMXX:1:2471:27561:29340 Basidiomycota (taxid 7825) 28|91 |:| 4:12 1:15 4:3 7825:2 0:25
下一步是使用sckmer脚本f分析各个barcodes 的k-mer统计信息。sckmer.r用于配对数据和sckmer_unpaired.r用于未配对/单端序列数据。这些函数通过barcode计算分配给一个分类单元的k-mers和唯一k-mers的数量。细胞barcode和UMI用于识别唯一条形码和reads。这里得到的结果报告了预先指定等级的分类群(默认属+种)的数据,并考虑了所有随后的更高分辨率等级。
Rscript sckmer.r --sample_name mytest --fa1 mytest/mytest_1.fa --fa2 mytest/mytest_2.fa --microbiome_output_file mytest/mytest.microbiome.output.txt --kraken_report mytest/mytest.kraken.report.txt --mpa_report mytest/mytest.kraken.report.mpa.txt --out_path mytest/ --cb_len 16 --umi_len 12
需要注意不同平台单细胞数据的barcode和UMI的长度/结构不同,这里的参数也不一样。然后我们就得到最关键的文件了,每个barcode对应的微生物注释信息。
$ head mytest/mytest.sckmer.txt
barcode taxid kmer uniq
CGATGGCGTAGCGTCC 28455 57 57
GTGCTTCAGACCATAA 28455 57 57
TTGCCTGCTGCCTTCC 28455 57 57
TTCGTTCATGATTAAT 7824 55 55
CTCTCTCTCTCTCCCC 8427 39 39
AACCAACACCTTTTCT 13684 57 57
AAGACAACAGATCACT 13684 114 57
AAGGTAAAGGAAGTAG 13684 114 57
ACATCGATCTAAGAAG 13684 114 59
接下来,就可以移步到Rstudio里面,来把这个文件和普通的单细胞表达矩阵结合在一起了。
# kraken report
report = read.delim('C:\\Users\\admin\\Desktop\\mytest\\mytest.kraken.report.txt', header = F)
report$V8 = trimws(report$V8)
report[report$V8 %in% c('Homo sapiens', 'Bacteria', 'Fungi', 'Viruses'), ]
head(report)
V1 V2 V3 V4 V5 V6 V7 V8
1 92.61 213559 213559 0 0 U 0 unclassified
2 7.39 17051 175 278367 5440 R 1 root
3 7.28 16798 2250 262335 5281 D 4 Eukaryota
4 6.17 14240 229 153814 3146 D1 6428 Opisthokonta
5 5.92 13652 0 145167 2979 D 6430 Holozoa
6 5.92 13652 51 145147 2973 K 12882 Metazoa (Animalia)
读入核心数据
# sckmer data
kmer_data = read.table('C:\\Users\\admin\\Desktop\\mytest\\mytest.sckmer.txt', header = T)
head(kmer_data)
barcode taxid kmer uniq
1 CGATGGCGTAGCGTCC 28455 57 57
2 GTGCTTCAGACCATAA 28455 57 57
3 TTGCCTGCTGCCTTCC 28455 57 57
4 TTCGTTCATGATTAAT 7824 55 55
5 CTCTCTCTCTCTCCCC 8427 39 39
6 AACCAACACCTTTTCT 13684 57 57
c = kmer_data %>%
subset(kmer > 1) %>%
group_by(taxid) %>%
mutate(nn = n()) %>%
subset(nn > 3) %>%
group_by(taxid)
c$name = report$V8[match(c$taxid, report$V7)] # add taxa names
head(c)
# A tibble: 6 × 6
# Groups: taxid [1]
barcode taxid kmer uniq nn name
<chr> <int> <int> <int> <int> <chr>
1 AACCAACACCTTTTCT 13684 57 57 65 Clavulicium
2 AAGACAACAGATCACT 13684 114 57 65 Clavulicium
3 AAGGTAAAGGAAGTAG 13684 114 57 65 Clavulicium
4 ACATCGATCTAAGAAG 13684 114 59 65 Clavulicium
5 ACGGTCGACTGCCGGC 13684 57 57 65 Clavulicium
6 ACGTAGTCATTAGGAA 13684 57 57 65 Clavulicium
读入单细胞转录组表达矩阵数据,并作标准化处理。
seo <- CreateSeuratObject(counts = datamy, project ="test", min.cells = 0, min.features = 0)
seo %>% NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData(features = rownames(seo)) %>%
RunPCA()%>%
FindNeighbors( dims = 1:50)%>%
FindClusters(resolution = 0.05)%>%
RunUMAP(dims = 1:10) ->seo
DimPlot(seo)
得到一张标准的UMAP图谱。
这个测试数据很小,不必在乎我的长相
把我们的微生物信息映射到UAMP上来。
paste0(c$barcode,"-1")
c$barcode_1 <- paste0(c$barcode,"-1")
head(c)
metaC <- as.data.frame(c)
head(metaC)
metaC$barcode <- metaC$barcode_1
row.names(metaC)<- metaC$barcode_1
# 数据格式整理,甚至还用了帮助文档
?AddMetaData
seo<- AddMetaData(seo,metadata = metaC )
head(seo@meta.data)
orig.ident nCount_RNA nFeature_RNA RNA_snn_res.0.05 seurat_clusters barcode
AAACCCAAGGAGAGTA-1 test 42 19 0 0 <NA>
AAACGCTTCAGCCCAG-1 test 41 29 0 0 <NA>
AAAGAACAGACGACTG-1 test 21 14 0 0 <NA>
AAAGAACCAATGGCAG-1 test 11 9 0 0 <NA>
AAAGAACGTCTGCAAT-1 test 29 19 0 0 <NA>
AAAGGATAGTAGACAT-1 test 36 22 0 0 <NA>
taxid kmer uniq nn name barcode_1
AAACCCAAGGAGAGTA-1 NA NA NA NA <NA> <NA>
AAACGCTTCAGCCCAG-1 NA NA NA NA <NA> <NA>
AAAGAACAGACGACTG-1 NA NA NA NA <NA> <NA>
AAAGAACCAATGGCAG-1 NA NA NA NA <NA> <NA>
AAAGAACGTCTGCAAT-1 NA NA NA NA <NA> <NA>
AAAGGATAGTAGACAT-1 NA NA NA NA <NA> <NA>
一张经典的映射上微生物丰度信息的UMAP就要映入眼帘了,你准备好了吗?
DimPlot(seo,group.by ='name' )+ Seurat::DarkTheme() + ggtitle("Single Cell Atlas \n mapping microbiome @SAHMI ")
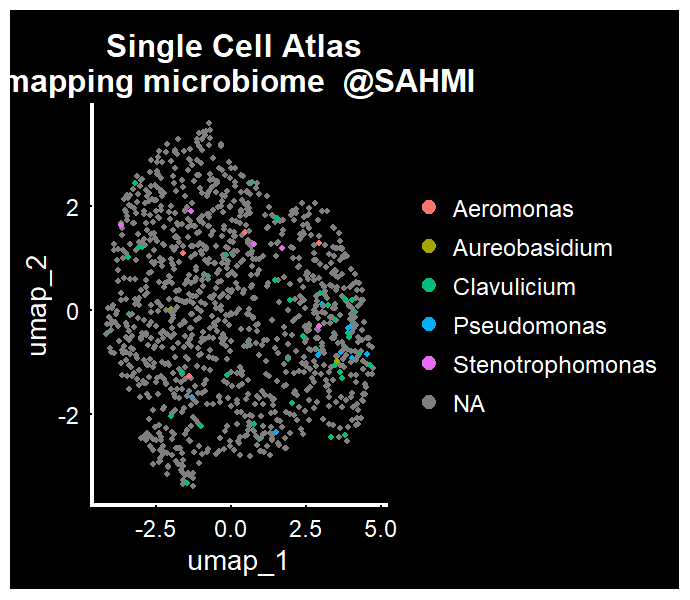
整个SAHMI分析到这里并没有结束,我建议仔细阅读原文档,并在github上与作者建立联系。有了这个对应关系,我们就可以在单细胞数据的框架里,细胞图谱、细胞轨迹、细胞互作等分析中,把微生物组的信息映射上去。也可以用微生物组的分析方法,在在亚群内或间分析微生物组的多样性,构建系统进化树等等,可分析的内容可是多了去了。
就像文中不时留下的问号一样,本文的测试也是有瑕疵的,欢迎大家勘误。不管怎么说,希望能给您的单细胞数据带来新的见解和可能性。做生物信息主要的一个自我感知,就是要知道生物信息学本身的局限性。那么,这里分析的单细胞微生物组在多大程度上是靠谱的?
- 单细胞转录组基于3'mRNA捕获,真的能够捕获到微生物的序列吗?
- 单细胞数据的产生,需要经历取样、组织解离、细胞分离、核酸捕获、建库,测序,而这些步骤哪些是无菌的?背景如何扣除?
- 作为基于生物信息学的方法,如何验证所得到的结果?
- 如何去掉假阳性?
本文代码和测试数据均为公开渠道获取,感兴趣的老师可以测试一下,并在这个过程中再次领略单细胞数据的无穷魅力。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-06-02,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号