单细胞5 拟时序分析
原创

1 拟时序分析
拟时序分析是为了探索自己感兴趣的几种细胞之间的发育关系,一般不是用全部类型的细胞来做的。实在不行问问ai,回答可详细
1.1 单样本拟时序分析
#rm(list = ls()) #单样本
library(Seurat)
library(monocle)
library(dplyr)
load("sce.Rdata") # 我真哭了,我要说一下,之前手动注释我跳过了,我说我怎么没有sce,在这研究了40分钟,终于让后我发现了问题所在,果然不能偷懒
> table(Idents(scRNA))
B_cell T_cells Monocyte Endothelial_cells
766 1645 127 158
Smooth_muscle_cells NK_cell
90 88
> head(scRNA@meta.data)
orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5
AAACCCACAGTCGGTC-1 SeuratProject 4243 1256 6.292717 1
AAACGAAAGAATCGCG-1 SeuratProject 7307 2577 2.572875 10
AAAGAACAGCTTACGT-1 SeuratProject 8154 1881 4.083885 1
AAAGAACAGGTTCATC-1 SeuratProject 8223 2182 4.377964 9
AAAGAACAGTCTGTAC-1 SeuratProject 3884 1377 4.763131 0
AAAGAACTCCACCTCA-1 SeuratProject 3997 1307 3.402552 3
seurat_clusters
AAACCCACAGTCGGTC-1 1
AAACGAAAGAATCGCG-1 10
AAAGAACAGCTTACGT-1 1
AAAGAACAGGTTCATC-1 9
AAAGAACAGTCTGTAC-1 0
AAAGAACTCCACCTCA-1 3
> DimPlot(scRNA,label = T)
> #输入数据是seurat做完降维聚类分群注释的数据。celltype是细胞类型注释,用以下代码添加
> scRNA$celltype = Idents(scRNA)
> #输入数据是seurat做完降维聚类分群注释的数据。celltype是细胞类型注释,用以下代码添加
> scRNA$celltype = Idents(scRNA) #做拟时序分析通常不是拿全部的细胞,而是拿感兴趣的一部分。用subset提取子集即可。
> #我的和花花老师的数据用的不太一样,因为跳过了手动注释,所以用的是自动注释的数据
> levels(Idents(scRNA)) #打出来细胞类型供复制
[1] "B_cell" "T_cells" "Monocyte" "Endothelial_cells"
[5] "Smooth_muscle_cells" "NK_cell"
> table(Idents(scRNA))
B_cell T_cells Monocyte Endothelial_cells
766 1645 127 158
Smooth_muscle_cells NK_cell
90 88
> scRNA = subset(scRNA,idents = c("NK_cell","T_cells"))#我就选这两个细胞吧
> table(Idents(scRNA))
T_cells NK_cell
1645 88
> # 以下两步进行抽样,实战跳过这步,实战可不能抽样。set.seed(1234) scRNA = subset(scRNA,downsample = 100)
> set.seed(1234)
> scRNA = subset(scRNA,downsample = 100)
>a = "sce_little.Rdata"
>save(scRNA,file = a)做一个小总结:拟时序分析可能并不是拿全部的细胞,而是一部分你自己想做的细胞,所以得提取子集,提取子集的函数是subset,可以用帮助文档查看使用方法。在做拟时序分析的时候,因为是采用差异基因进行排序的,所以要求是两类细胞或者两类以上(要选择的细胞亲缘关系要近一点,有分化的可能性,完全不挨着的细胞不太行)。如果非得只想做一类细胞的话,有两种方法1把这类细胞分群,2选择其他基因
有一个需要注意的问题,monocle和seurat两个包不互通,所以需要将seurat对象转换为monocle可以接受的CellDataSet对象,理论上,importCDS函数可以直接把seurat对象转换为CellDataSet对象,但是monocle和seurat,更新速度不一样,所以目前不行。
1.2 创建CellDataSet对象
> # count矩阵,官方建议用count
> ct <- scRNA@assays$RNA$counts #提取count
> # 基因注释
> gene_ann <- data.frame(
+ gene_short_name = row.names(ct),
+ row.names = row.names(ct)
+ )
> fd <- new("AnnotatedDataFrame",
+ data=gene_ann)
> # 临床信息
> pd <- new("AnnotatedDataFrame",
+ data=scRNA@meta.data)
> #新建CellDataSet对象
> sc_cds <- newCellDataSet(
+ ct,
+ phenoData = pd,
+ featureData =fd,
+ expressionFamily = negbinomial.size(),
+ lowerDetectionLimit=1)
> sc_cds
CellDataSet (storageMode: environment)
assayData: 20648 features, 188 samples
element names: exprs
protocolData: none
phenoData
sampleNames: AAAGAACAGGTTCATC-1 AACAACCTCGTTTACT-1 ... TTTGGTTAGCTAATCC-1 (188
total)
varLabels: orig.ident nCount_RNA ... Size_Factor (8 total)
varMetadata: labelDescription
featureData
featureNames: AL627309.1 AL627309.3 ... AC240274.1 (20648 total)
fvarLabels: gene_short_name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: 1.3 构建细胞发育轨迹
在这个过程要选择基因,不同手段
1.使用seurat给出的高变化基因
2.按照平均表达量大于某个数字(比如0.1,官网用的是这个)的基因。
3.使用不同细胞类型之间的差异基因,differentialGeneTest计算(这个是最好的)。
个人理解,为什么要使用不同细胞间的差异基因呢,因为确实是没有差异怎么看不同,怎么推到分化过程。
差异分析十分漫长,耗费电脑资源,所以提前看好多少个核,空几个剩下全用

我的电脑cpu
#构建系统发育轨迹
sc_cds <- estimateSizeFactors(sc_cds)
sc_cds <- estimateDispersions(sc_cds)
fdif = "diff_test_res.Rdata"
if(!file.exists(fdif)){
diff_test_res <- differentialGeneTest(sc_cds,
fullModelFormulaStr = "~celltype",
cores = 10) #16个cpu用10个
save(diff_test_res,file = fdif)
}
load(fdif)
ordering_genes <- row.names(subset(diff_test_res, qval < 0.01))
#查看基因,筛选适合用于排序的,设置为排序要使用的基因
head(ordering_genes)
#[1] "TNFRSF18" "TNFRSF9" "MRTO4" "RUNX3" "CD52" "FGR"
length(ordering_genes)
sc_cds <- setOrderingFilter(sc_cds, ordering_genes)
#画出选择的基因
plot_ordering_genes(sc_cds)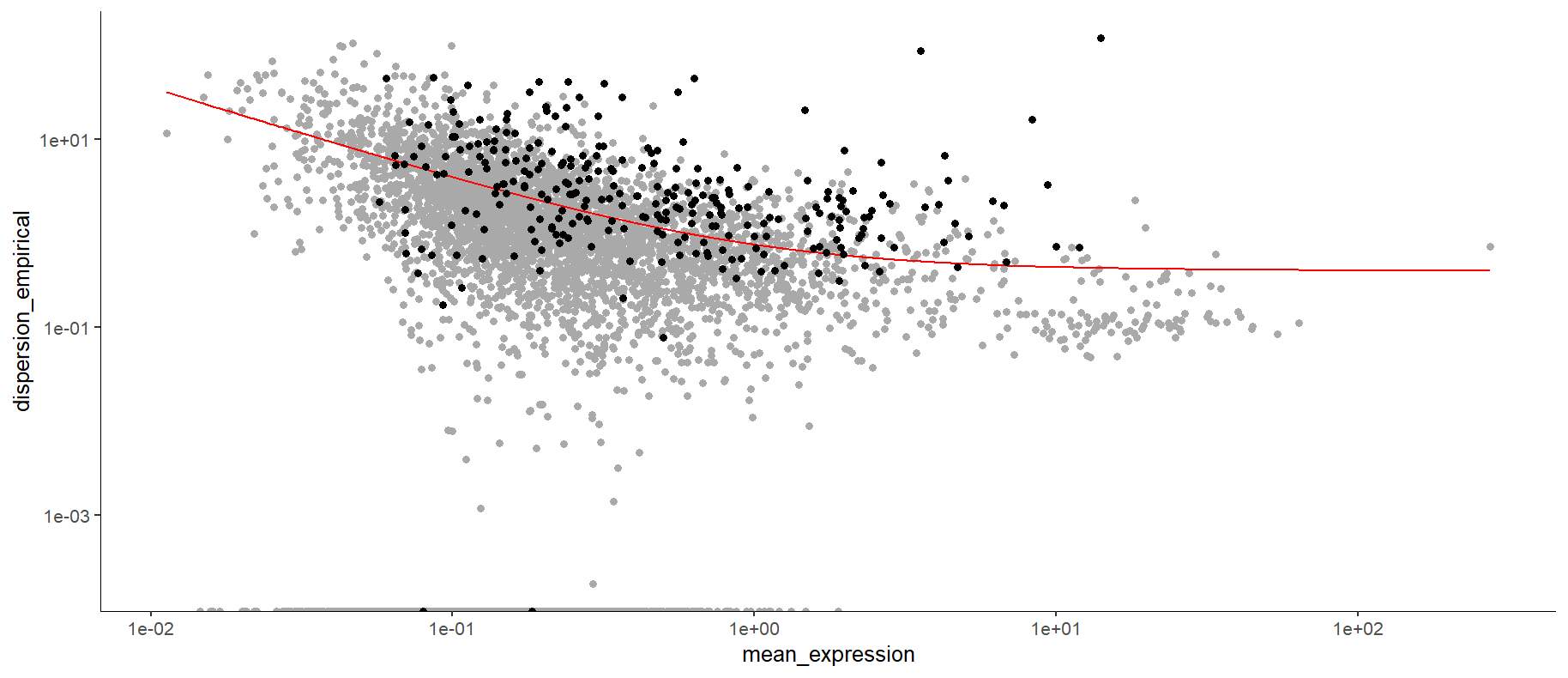
细胞发育轨迹图
这个轨迹图中黑色的点就是我们要选择的基因,这个选择方式有很多,有时候可能是红线右上角。
这个细胞发育轨迹图,plot_ordering_genes画的图纵坐标是基因表达量的变异性,,横坐标是每个基因在所有细胞种的平均表达量。(有些教程选择的是变异性强,表达量高的一些基因)
之后要进行降维,和排序,排序是要模拟细胞从一个状态逐渐发展到立一个状态的过程
sc_cds <- reduceDimension(sc_cds)#降维,理解
sc_cds <- orderCells(sc_cds)#细胞排序,拟时序分析假设细胞状态的变化是连续的,通过排序可以模拟细胞从一个状态逐渐发展到另一个状态的过程,这样才方便推算分化过程。1.4绘图展示
1.4.1发育轨迹图
library(ggsci) #需要用到这个包
p1 = plot_cell_trajectory(sc_cds)+ scale_color_nejm()
p1 #表示了细胞从一个状态到另一个状态的转变过程
p2 = plot_cell_trajectory(sc_cds, color_by = 'Pseudotime')
p2 #伪时间(Pseudotime)图,可能代表细胞分化过程
p3 = plot_cell_trajectory(sc_cds, color_by = 'celltype') + scale_color_npg()
p3 #细胞轨迹图根据细胞类型着色
library(patchwork)
p2+p1/p3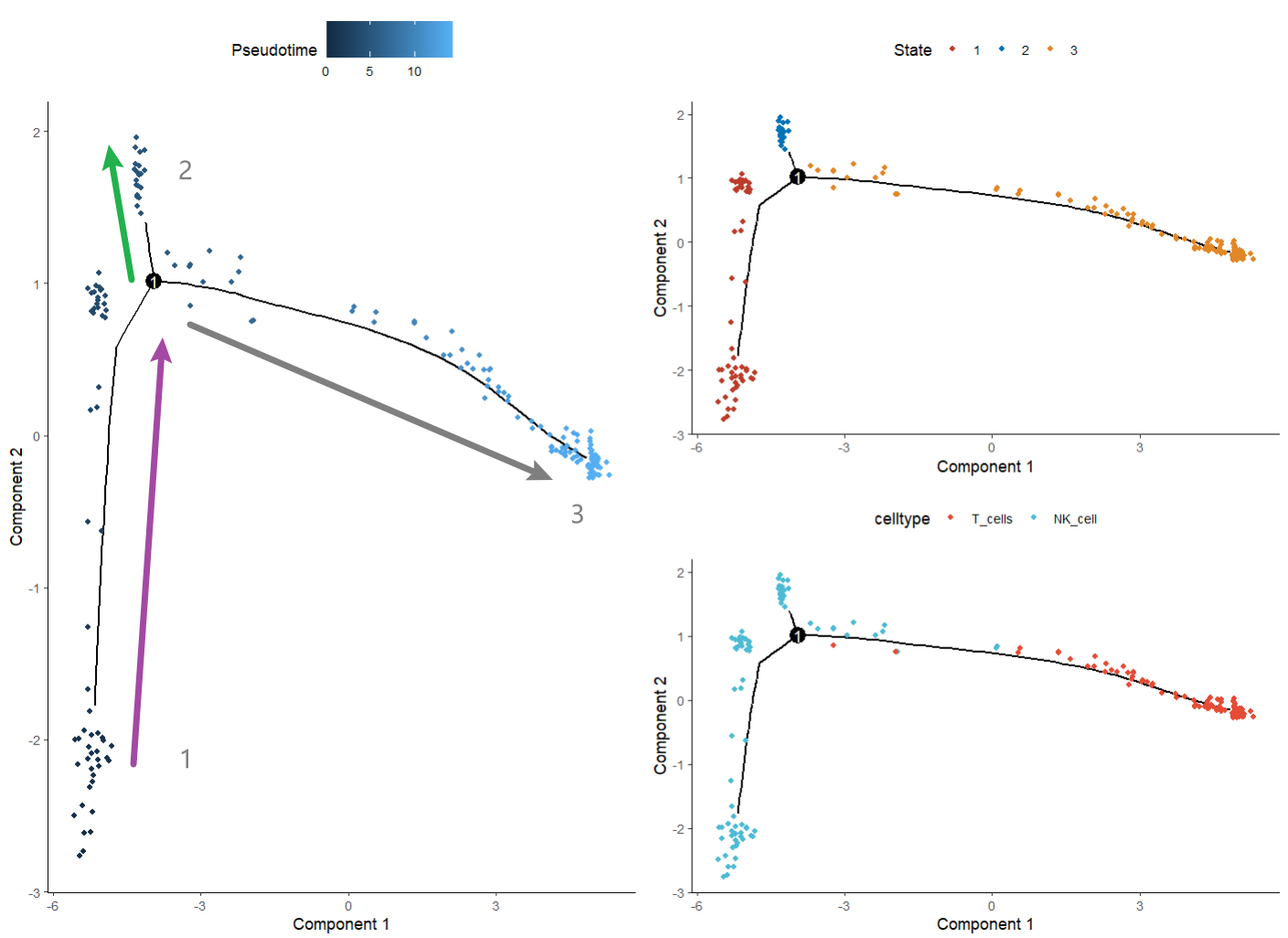
Pseudotime数值从小到大就是顺序就是推断的发育顺序。图上的点颜色越深,时间越早,颜色越浅,时间越晚。可以理解为分化关系我觉得
state是发育的不同阶段,数值越小越靠前。这个就是从早期到晚期的一个过程
celltype则可以看到具体的细胞类型在时间轨迹图上的分布。大致分析一下Tcells是成熟细胞了,NK细胞会发育成为两种状态
1.4.2 经典的拟时序分析
展示了一些基因是如何随着时间轨迹的变化而变化的,体现变化过程,选择q值小的(是不是忘了为啥,q值是错误值相当于,越小越可靠。
> gene_to_cluster = diff_test_res %>% arrange(qval) %>% head(50) %>% pull(gene_short_name);head(gene_to_cluster)
[1] "GNLY" "IGKC" "XCL1" "AREG" "KLRD1" "TYROBP"
> #arrange(qval)排序
> plot_pseudotime_heatmap(sc_cds[gene_to_cluster,],
+ num_clusters = nlevels(Idents(scRNA)),
+ show_rownames = TRUE,
+ cores = 4,return_heatmap = TRUE,
+ hmcols = colorRampPalette(c("navy", "white", "firebrick3"))(100))
1.4.3 基因轨迹图
用感兴趣的基因给轨迹图着色,可以体现某一个基因随着轨迹的表达情况变化,(基因表达水平在细胞发展过程中的变化趋势)
gs = head(gene_to_cluster) #基因可以换成你感兴趣的啊
plot_cell_trajectory(sc_cds,markers=gs,
use_color_gradient=T)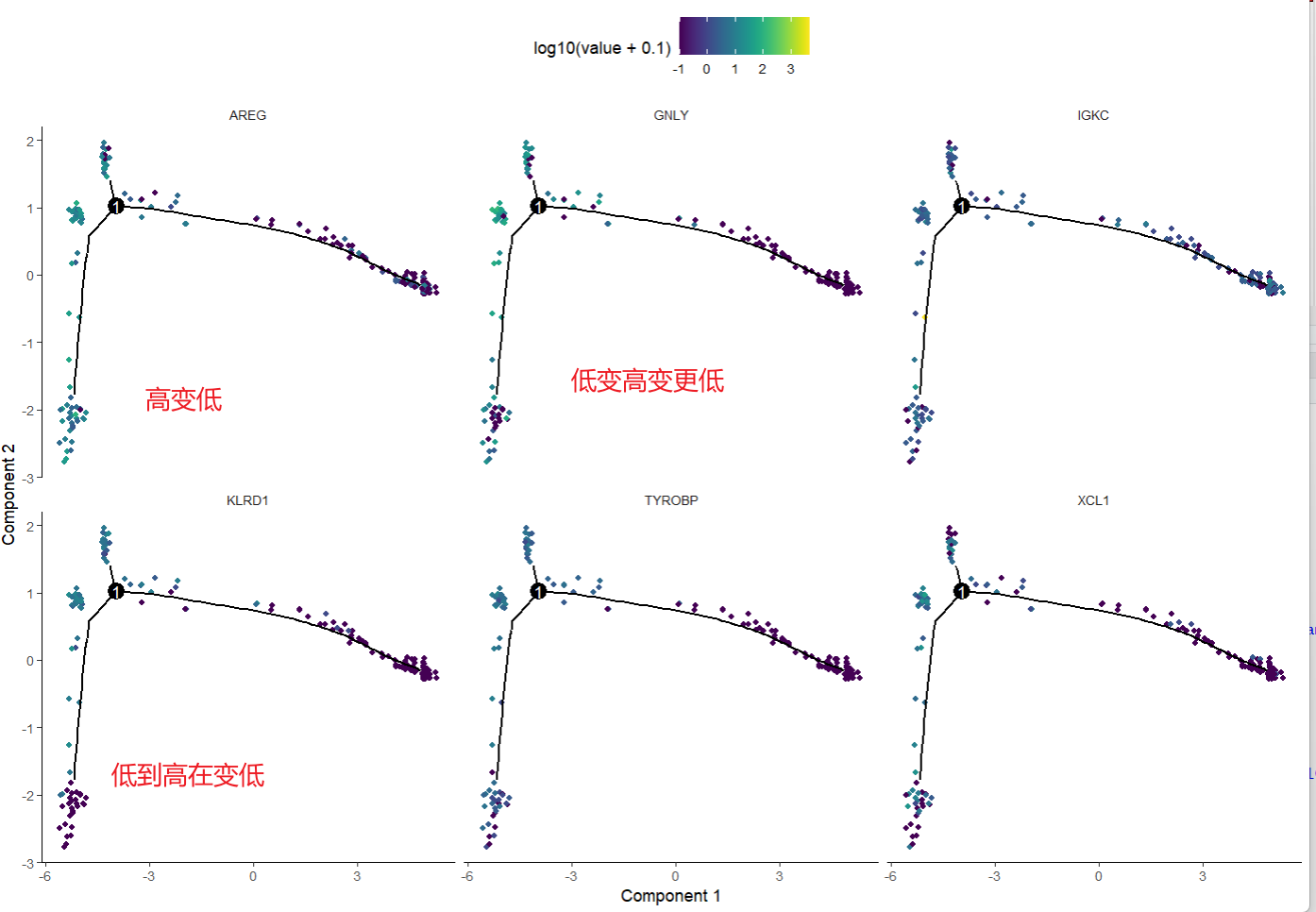
有一些变化不明显
1.4.3 基因拟时序点图
基因表达水平随拟时序变化的可视化方法
plot_genes_in_pseudotime(sc_cds[gs,], #基因拟时序点图
color_by = "celltype",
nrow= 3, #6个基因所以排了3行,数量有变化时要改
ncol = NULL )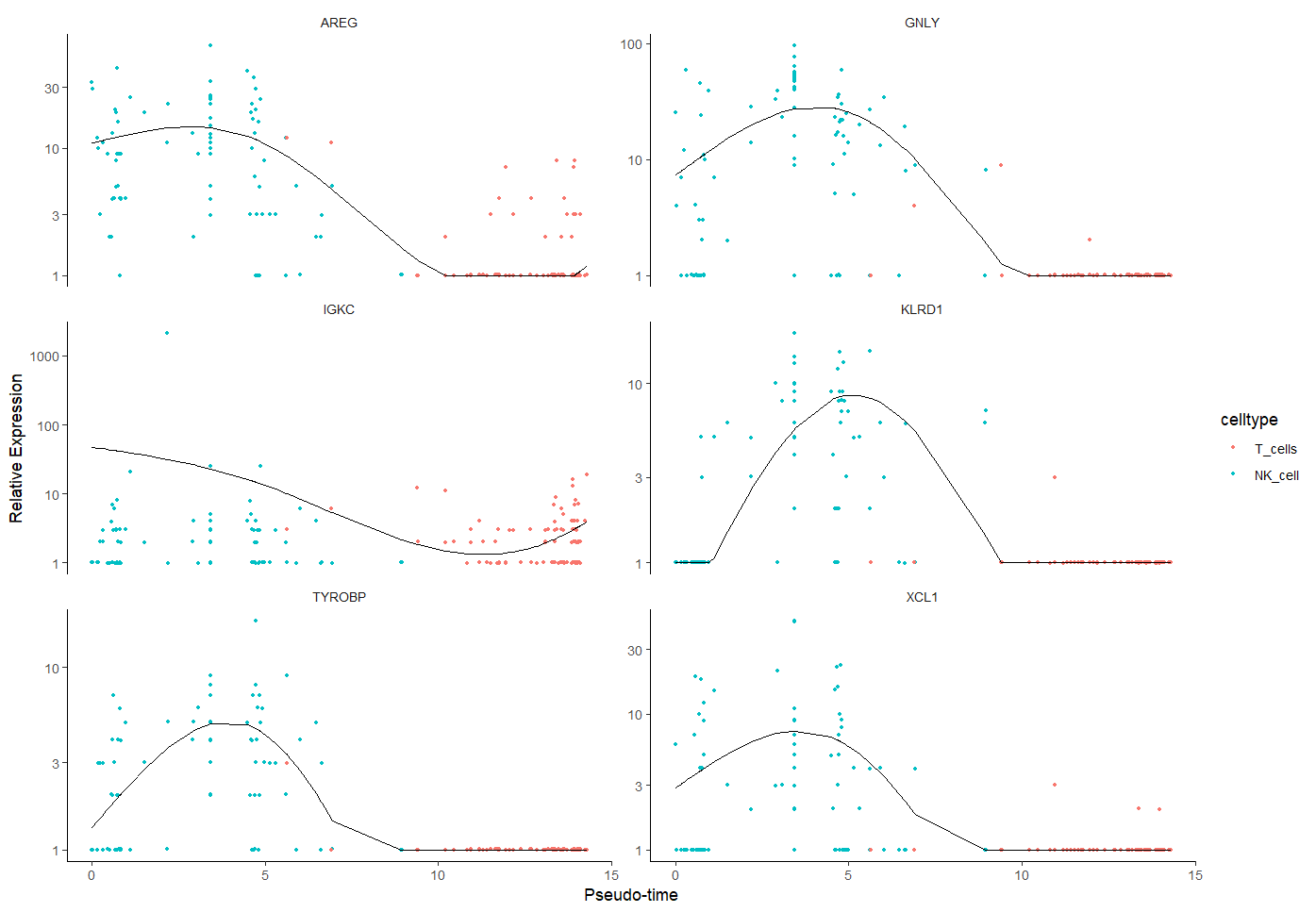
基因随着拟时序的变化趋势,趋势线有
2 多样本的拟时序
多样本相当于批量处理,就是直接把不同样本的区别一下
head(scRNA@meta.data) #多样本,scRNA是多样本的哪个sce.all
rm(list = ls())
library(Seurat)
library(monocle)
library(dplyr)
load("scRNA.Rdata")
table(Idents(scRNA)) #计数
table(scRNA$orig.ident) #计数
head(scRNA@meta.data)
DimPlot(scRNA,label = T)+NoLegend() #画图
scRNA$celltype = Idents(scRNA)
levels(Idents(scRNA)) #打出来细胞类型供复制
scRNA = subset(scRNA,idents = c("CD8+ T-cells","NK cells"))
table(Idents(scRNA))
#set.seed(1234) #抽样,实战不行
#scRNA = subset(scRNA,downsample = 100) #抽样,实战不行
table(Idents(scRNA))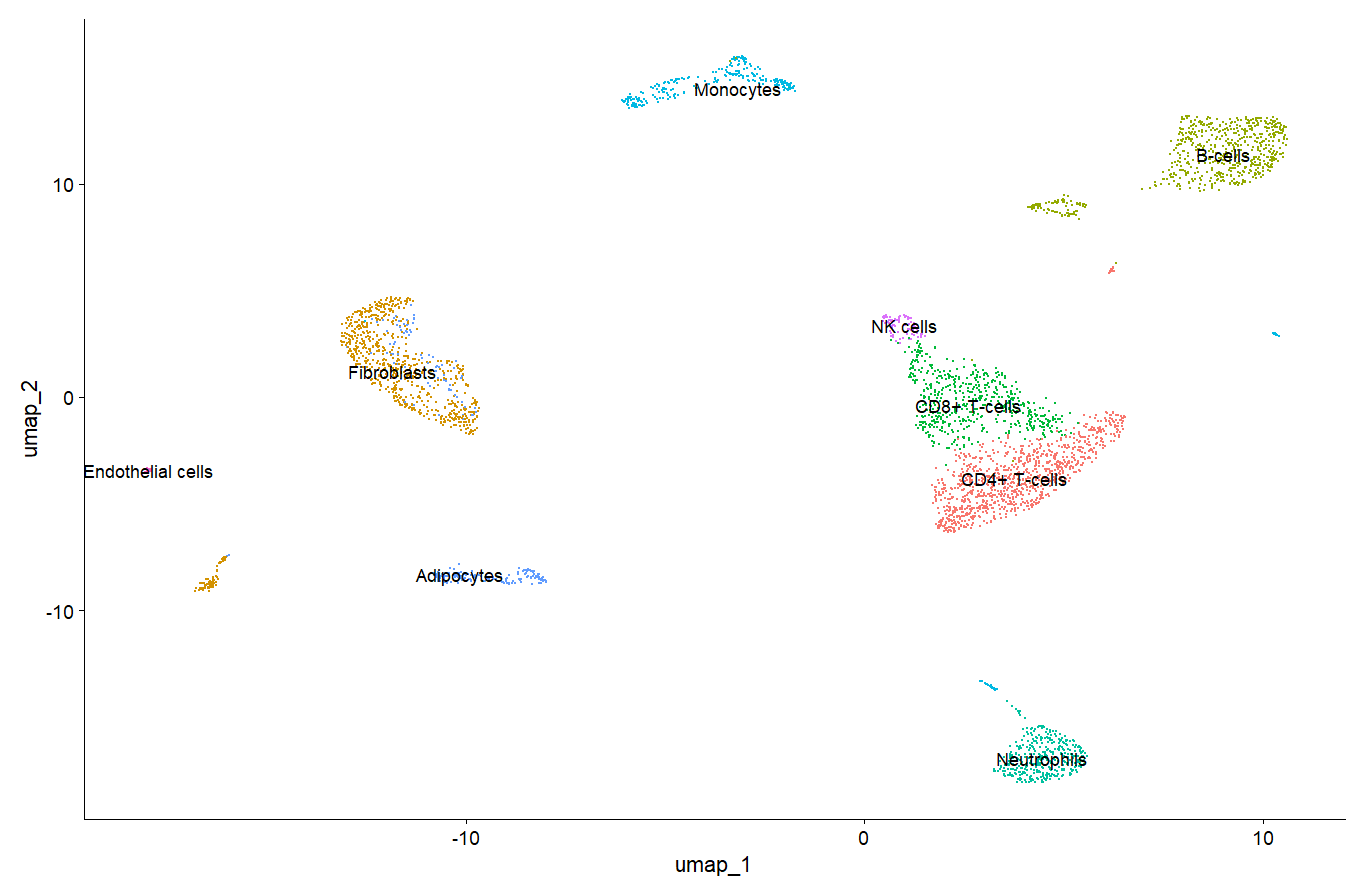
2.2 创建CellDataSet对象
CellDataSet对象是Scanpy库中用于存储和处理单细胞数据的一种数据结构
> # count矩阵,官方建议用count
> ct <- scRNA@assays$RNA$counts
> # 基因注释
> gene_ann <- data.frame(
+ gene_short_name = row.names(ct),
+ row.names = row.names(ct)
+ )
> fd <- new("AnnotatedDataFrame",
+ data=gene_ann)
> # 临床信息
> pd <- new("AnnotatedDataFrame",
+ data=scRNA@meta.data)
> #新建CellDataSet对象
> sc_cds <- newCellDataSet(
+ ct,
+ phenoData = pd,
+ featureData =fd,
+ expressionFamily = negbinomial.size(),
+ lowerDetectionLimit=1)
> sc_cds
CellDataSet (storageMode: environment)
assayData: 24357 features, 187 samples
element names: exprs
protocolData: none
phenoData
sampleNames: ACCTGAACAGACCTGC-1_1 AGAAATGAGTTGTCAC-1_1 ...
TTTGTTGGTCCAGGTC-1_6 (187 total)
varLabels: orig.ident nCount_RNA ... Size_Factor (10 total)
varMetadata: labelDescription
featureData
featureNames: AL627309.1 AL627309.3 ... AF127577.3 (24357 total)
fvarLabels: gene_short_name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: 2.3 构建细胞发育轨迹
> sc_cds <- estimateSizeFactors(sc_cds)
> sc_cds <- estimateDispersions(sc_cds)
Removing 97 outliers
> fdif = "diff_test_res2.Rdata"
> if(!file.exists(fdif)){
+ diff_test_res <- differentialGeneTest(sc_cds,
+ fullModelFormulaStr = " ~ celltype + orig.ident",
+ reducedModelFormulaStr = " ~ orig.ident",
+ cores = 8)
+ save(diff_test_res,file = fdif)
+ }
> load(fdif)
> ordering_genes <- row.names(subset(diff_test_res, qval < 0.01))
> #查看基因,筛选适合用于排序的,设置为排序要使用的基因
> head(ordering_genes)
[1] "EFHD2" "RCAN3" "JUN" "LMNA" "FCRL6" "SLAMF1"
> length(ordering_genes)
[1] 116
> sc_cds <- setOrderingFilter(sc_cds, ordering_genes)
> #画出选择的基因
> plot_ordering_genes(sc_cds)
> #降维
> sc_cds <- reduceDimension(sc_cds,residualModelFormulaStr = "~orig.ident") #这块用的是reduceDimension不太一样了,而且需要重新去除批次效应
Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
Also defined by 'spam'
Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
Also defined by 'spam'
> #细胞排序
> sc_cds <- orderCells(sc_cds)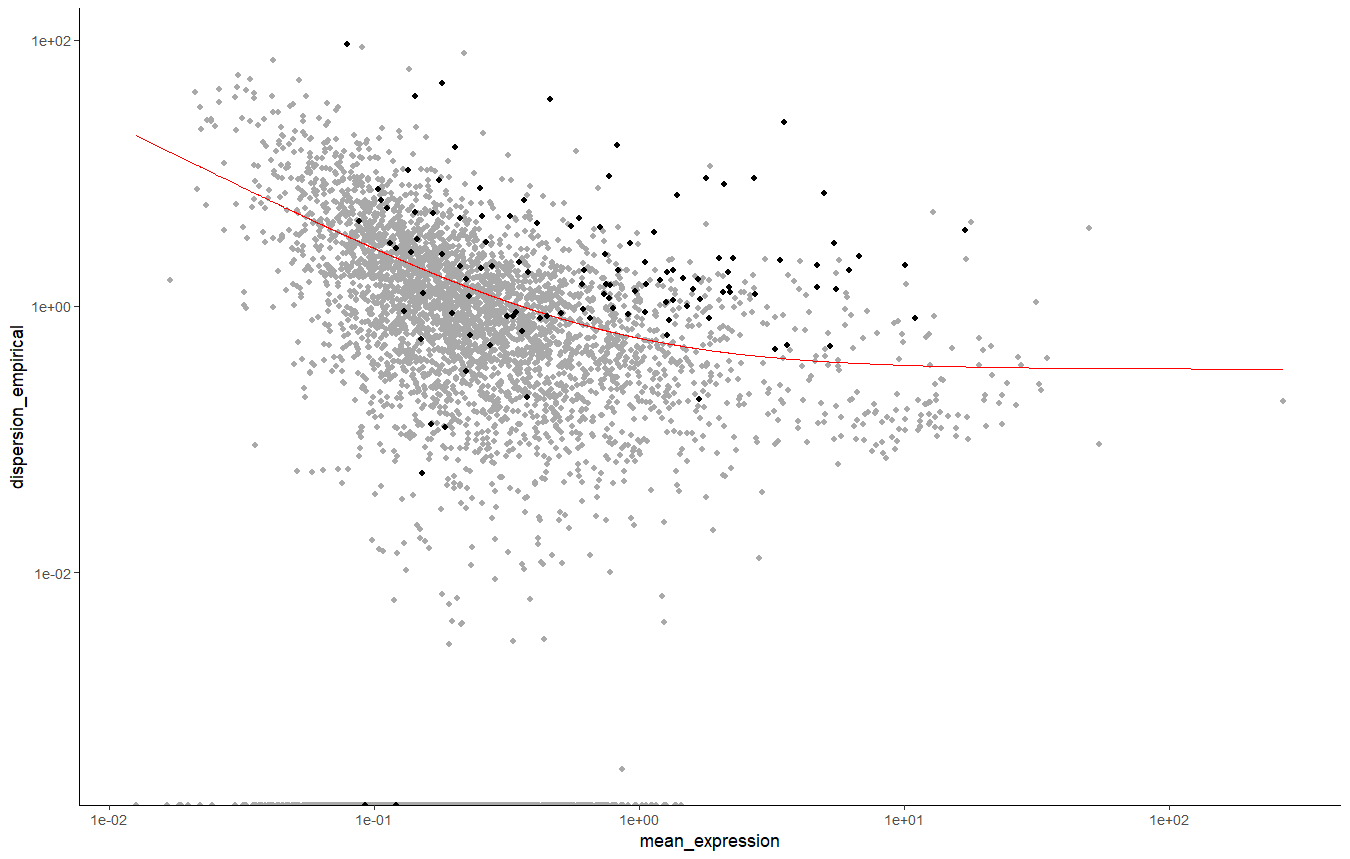
2.4 绘图
2.4.1 发育轨迹图
library(ggsci)
p1 = plot_cell_trajectory(sc_cds)+ scale_color_nejm()
p2 = plot_cell_trajectory(sc_cds, color_by = 'Pseudotime')
p3 = plot_cell_trajectory(sc_cds, color_by = 'celltype') + scale_color_npg()
library(patchwork)
p2+p1/p3
plot_cell_trajectory(sc_cds, color_by = 'orig.ident') #不同样本的一个细胞分布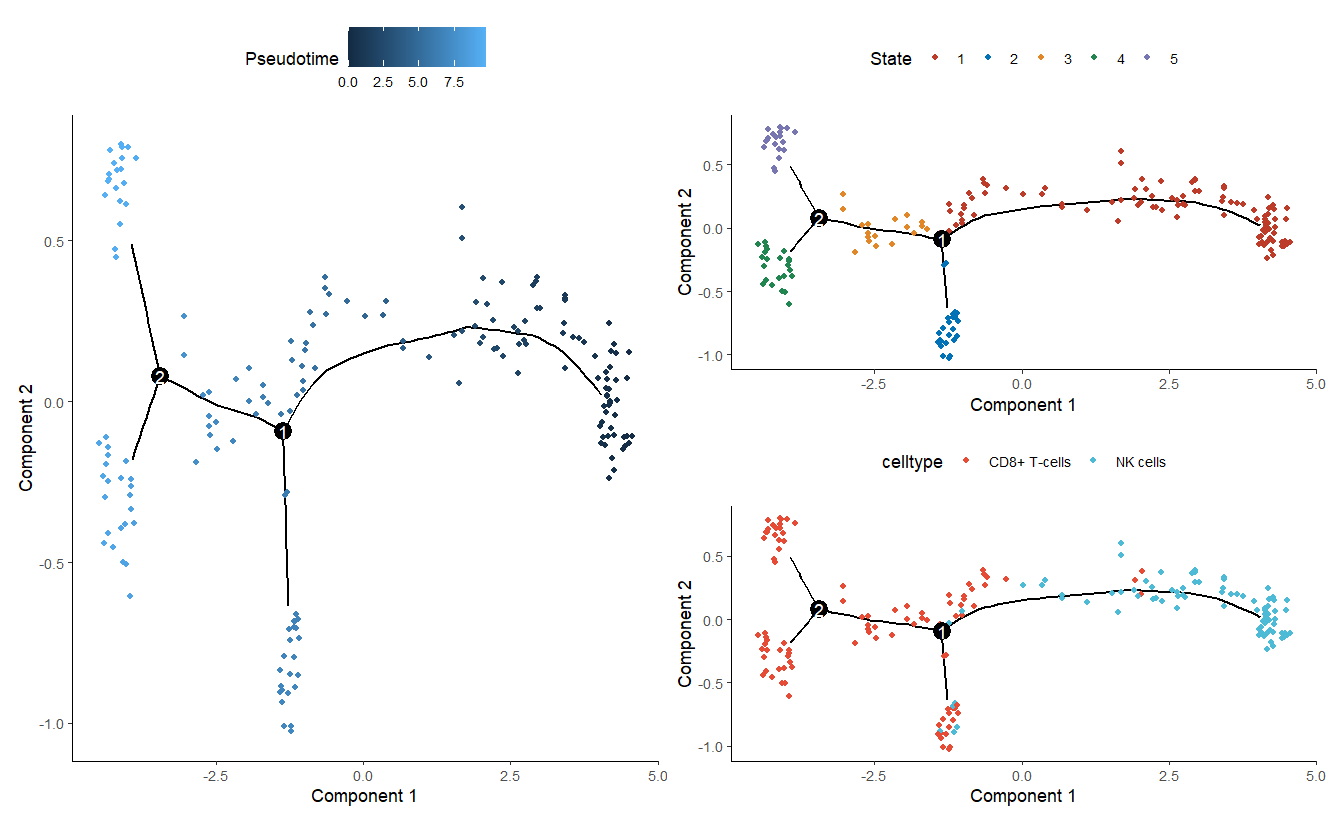
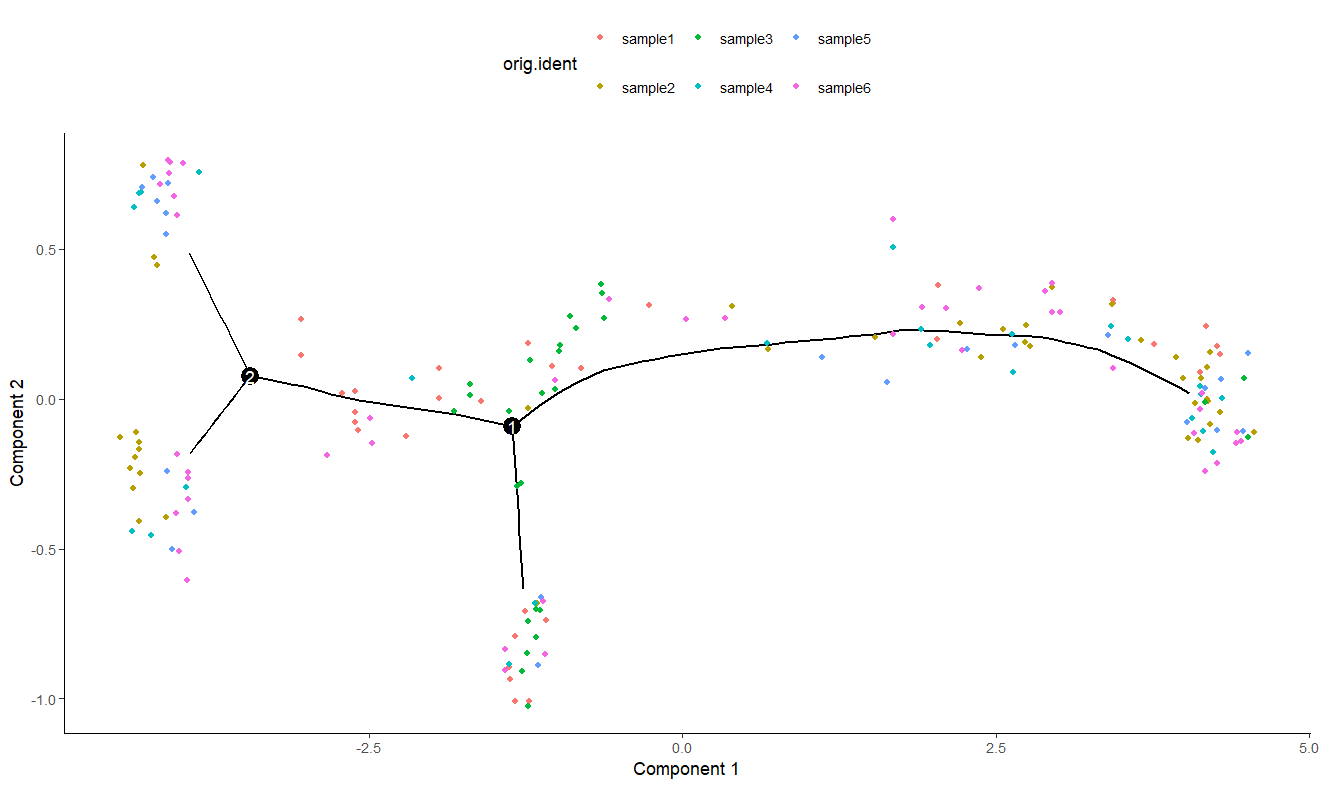
没有说一个样本在某一块聚集了,证明去除了批次效应
2.4.2 拟时序热图
> gene_to_cluster = diff_test_res %>% arrange(qval) %>% head(50) %>% pull(gene_short_name);head(gene_to_cluster)
[1] "GNLY" "GZMB" "FGFBP2" "NKG7" "CCL4" "TYROBP"
> plot_pseudotime_heatmap(sc_cds[gene_to_cluster,],
+ num_clusters = nlevels(Idents(scRNA)),
+ show_rownames = TRUE,
+ cores = 4,return_heatmap = TRUE,
+ hmcols = colorRampPalette(c("navy", "white", "firebrick3"))(100))
2.4.3 基因轨迹图
gs = head(gene_to_cluster) #gs可以换成你想换的基因
plot_cell_trajectory(sc_cds,markers=gs,
use_color_gradient=T)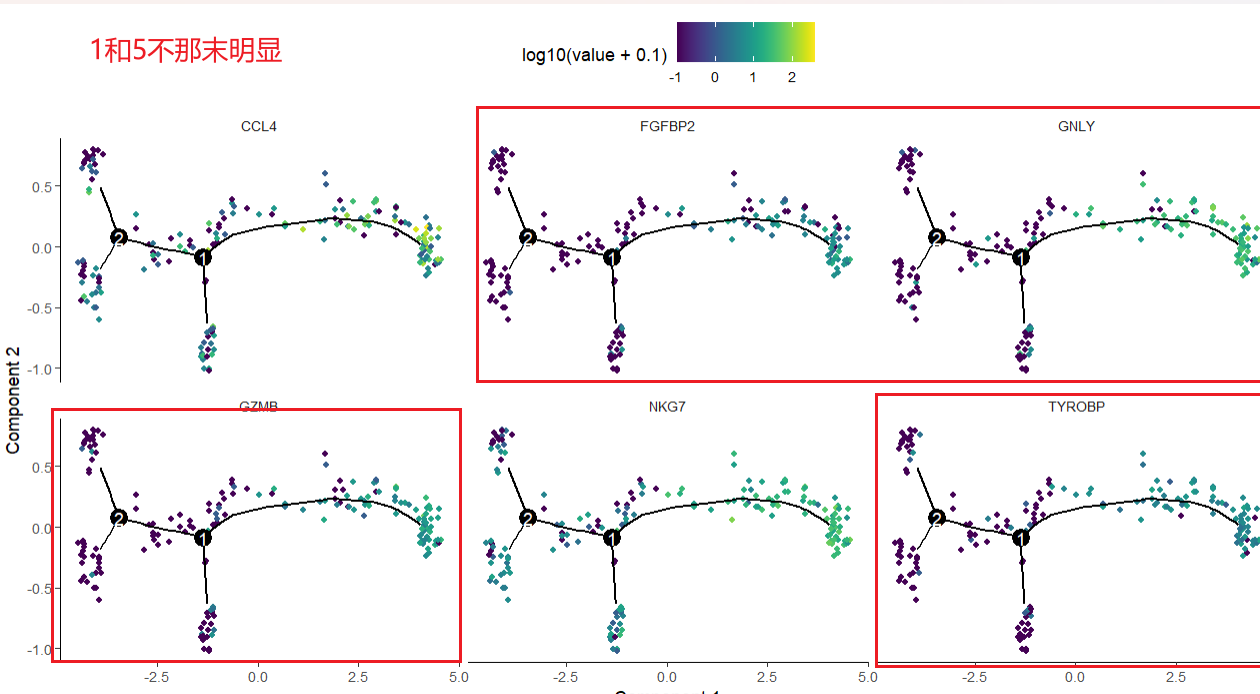
2.4.4 基因拟时序点图
横坐标是按照pseudotime排好顺序的。
plot_genes_in_pseudotime(sc_cds[gs,],
color_by = "celltype",
nrow= 2, #6个基因所以排2行看看,数量有变化时要改
ncol = NULL )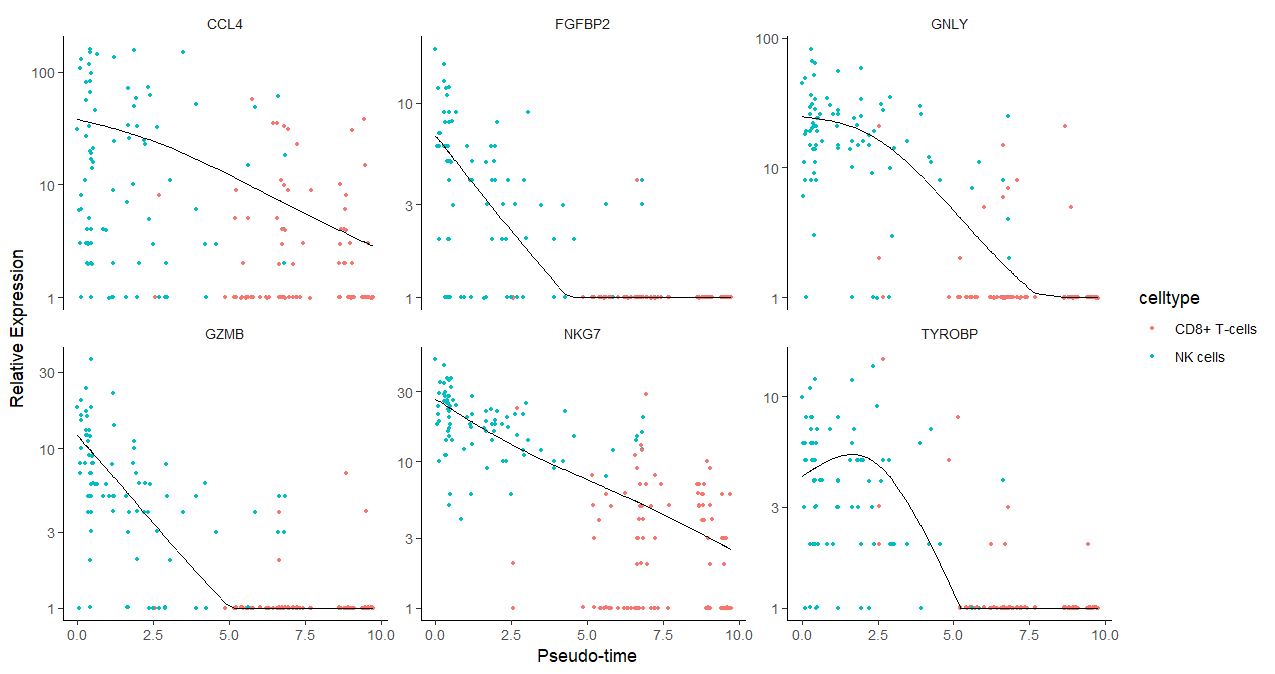
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号