单细胞测序—不同格式的单细胞测序数据读写(多样本)
原创
单细胞测序—不同格式的单细胞测序数据读写(多样本)
原创
sheldor没耳朵
发布于 2024-08-25 12:53:13
发布于 2024-08-25 12:53:13
单细胞测序—不同格式的单细胞测序数据读写(多样本)
这里记录下不同格式的单细胞测序数据读写,存在5种常见的单细胞测序数据。
读写过程中需要将一个GSE数据集中多个样本的seurat对象合并成一个大的seurat对象
1 10X标准格式
1.1 10X数据读取
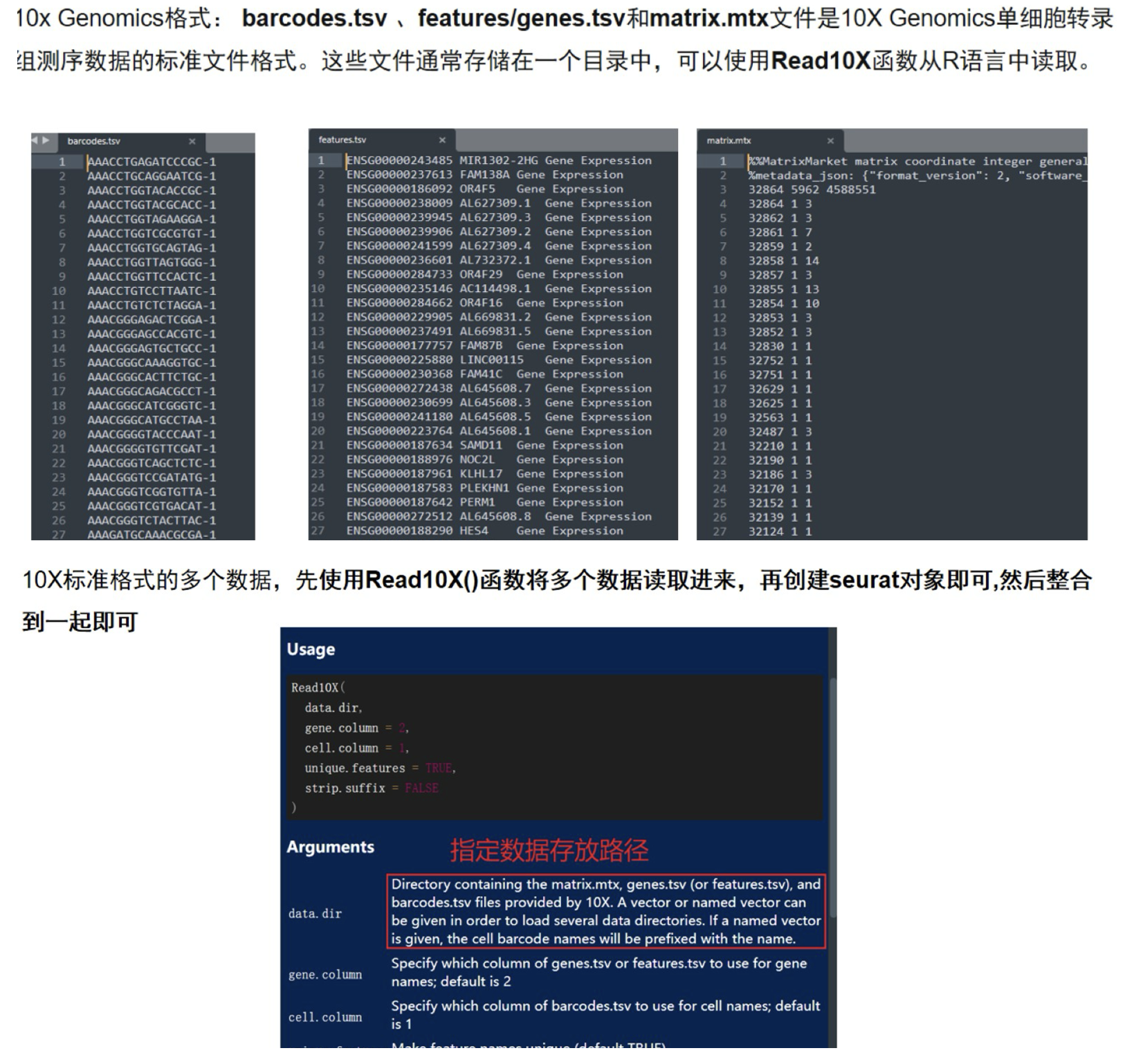
#清空环境 加载需要的R包
rm(list=ls())
options(stringsAsFactors = F)
source('./lib.R')
##10X标准格式
dir='GSE212975_10x/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident) 注:lib.R
library(COSG)
library(harmony)
library(ggsci)
library(dplyr)
library(future)
library(Seurat)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(ggplot2)
library(patchwork)
library(stringr)1.2 代码解释
sceList = lapply(samples,function(pro){
if(...)
}通过遍历一个样本列表,将每个样本的原始数据文件加载到R中,然后创建一个Seurat对象,最后将所有Seurat对象存储在一个列表 (sceList) 中。
- Read10X(file.path(dir, pro)):file.path(dir, pro) 将目录路径 dir 和当前样本文件名 pro 拼接成一个完整的文件路径。
- 这里的 if语句检查 tmp
是否包含两个数据层:
- if(length(tmp) == 2):如果 tmp 的长度为2,说明它包含两个不同的数据层(如gene expression和 protein expression),则选择第一个数据层(通常是基因表达数据 tmp[1])。
- else { ct = tmp }:如果 tmp的长度不是2,那么直接将 tmp赋值给 ct。在这种情况下,ct 包含的是单层数据,如基因表达矩阵。
do.call(rbind, lapply(sceList, dim))- lapply(sceList, dim):lapply 函数遍历 sceList中的每个Seurat对象,并对每个对象应用 dim函数,返回每个对象的维度(即基因数和细胞数)。
- do.call(rbind, ...):do.call 函数将 lapply 返回的结果(每个对象的维度)按行绑定(rbind),生成一个矩阵,矩阵的每一行对应一个样本的数据维度。这个矩阵便于查看每个样本的基因数和细胞数。
sce.all = merge(x = sceList[[1]],
y = sceList[-1],
add.cell.ids = samples)merge函数用于将多个Seurat对象合并为一个大的Seurat对象。
- x = sceList[1]:指定第一个Seurat对象作为合并的基础。
- y = sceList-1:合并列表中其余的Seurat对象。sceList-1表示 sceList列表中除了第一个对象以外的所有对象。
- add.cell.ids = samples:为每个样本的细胞添加唯一的标识符,这样在合并后可以区分不同样本的细胞。samples 是样本名称的列表,这些名称将作为每个样本细胞的前缀。
合并后,sce.all 是一个包含所有样本的单个Seurat对象,包含所有细胞的基因表达数据。
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)- names(sce.all@assays$RNA@layers):返回 sce.all对象中 RNA 这个assay中的所有数据层的名称。Seurat对象可以包含多个数据层(如 counts、data、scale.data),不同的数据层表示数据在不同处理阶段的信息。
- sce.all["RNA"]$counts:直接访问Seurat对象中 RNA assay 的 counts 数据层,这个数据层通常包含的是原始的未标准化的基因表达计数矩阵。
- LayerData(sce.all, assay = "RNA", layer = "counts"):这是另一种访问 RNA assay 中 counts数据层的方法。这个函数的功能与上面的直接访问方法相同,但可以在代码中显式指定你想访问的assay和数据层,更加灵活。
- JoinLayers(sce.all):将 sce.all 对象中的不同数据层进行合并,通常是为了将处理后的数据层与原始数据层同步。例如,处理后的表达矩阵(data 层)和原始计数矩阵(counts层)可能会合并,确保对象中的所有数据层都包含相同的细胞和基因集合。 在 Seurat 中,一个 Seurat 对象通常包含多个数据层(layers),如: counts: 原始的未处理的基因表达计数。 data: 经过标准化的表达数据。 scale.data: 经过缩放处理的数据,用于下游分析(如PCA、聚类等)。
这些数据层在Seurat对象的assay中存储,通常命名为 "RNA"。
JoinLayers 是 Seurat 中的一个辅助函数,用来确保 Seurat 对象中所有数据层(如 counts、data、scale.data)包含相同的基因和细胞。如果你对某些层进行了操作,比如过滤掉了一些基因或细胞,而没有对其他层进行相同的操作,JoinLayers 会通过同步这些层来修复这个不一致性。
换句话说,JoinLayers 会对所有数据层进行检查,并确保它们的维度(基因数和细胞数)一致。如果有任何层在之前的操作中缺失了某些基因或细胞,JoinLayers 会根据现有的层来补全。
im(sce.all[["RNA"]]$counts)
table(sce.all$orig.ident)- 检查
sce.all对象中RNAassay 的counts数据层的维度。在合并多个 Seurat 对象之后,确认最终合并后的对象包含的基因数量和细胞数量。 - 对
orig.ident进行计数,生成每个样本中细胞数量的频率表。统计每个样本贡献的细胞数量,确认数据的分布情况。
1.3 补充:GEO下载数据整理脚本
如在GEO下载测序数据时候,我们需要进行初步的数据整理,即将每个样本的三个数据文件(barcode\features\matrix)整理在各自的文件夹中,并规范命名。
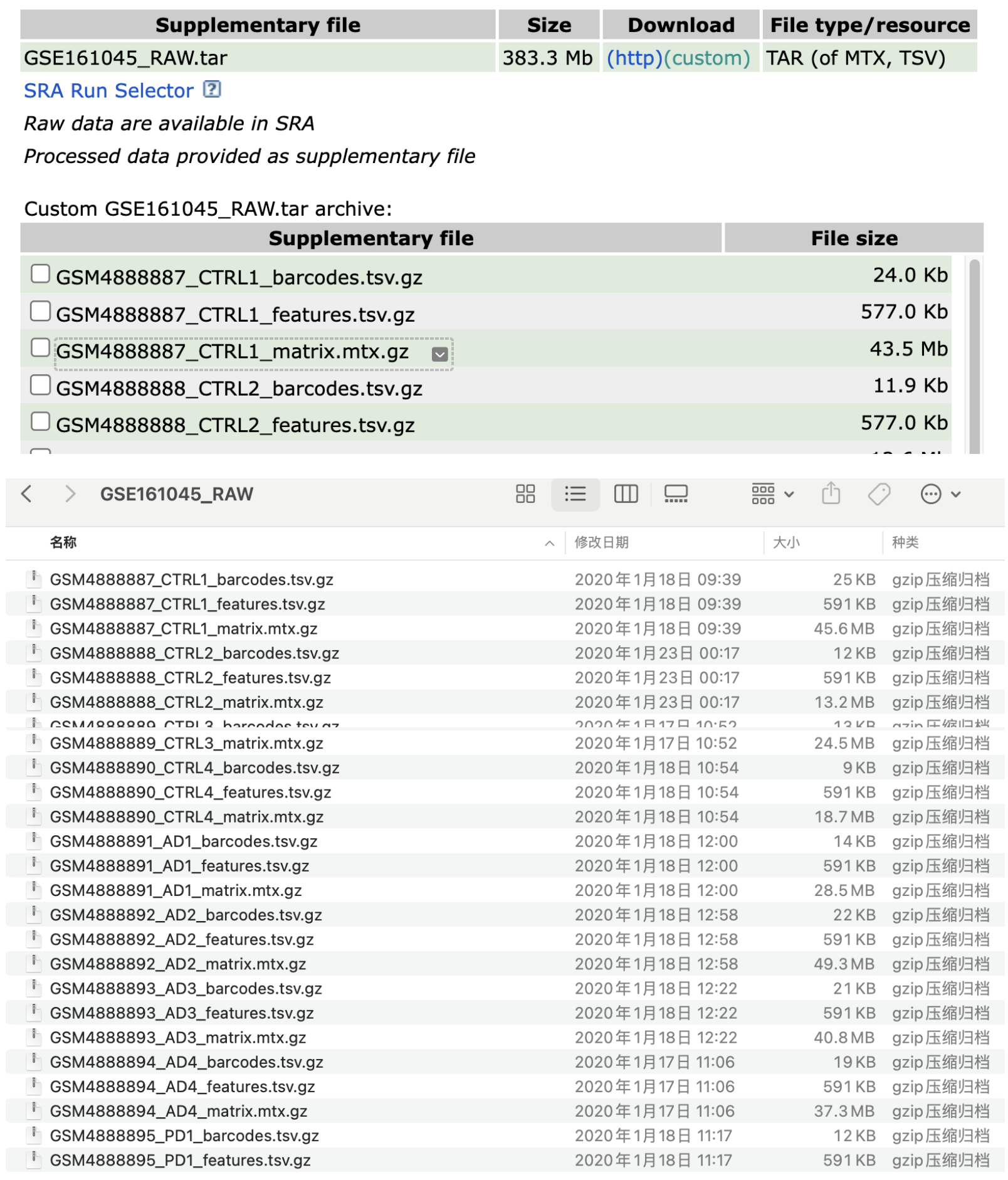
fs=list.files('./','features')
fs
samples= gsub('_features.tsv.gz','',fs)
samples
lapply(1:length(samples), function(i){
x=samples[i]
y=fs[i]
dir.create(x,recursive = T)
file.copy(from= y ,
to=file.path(x, 'features.tsv.gz' ))
file.copy(from= gsub('features','matrix',gsub('tsv','mtx',y)),
to= file.path(x, 'matrix.mtx.gz' ) )
file.copy(from= gsub('features','barcodes',y),
to= file.path(x, 'barcodes.tsv.gz' ))
})2 h5格式
h5格式在一个文件里同时包括了feature、bacode、matrix的信息
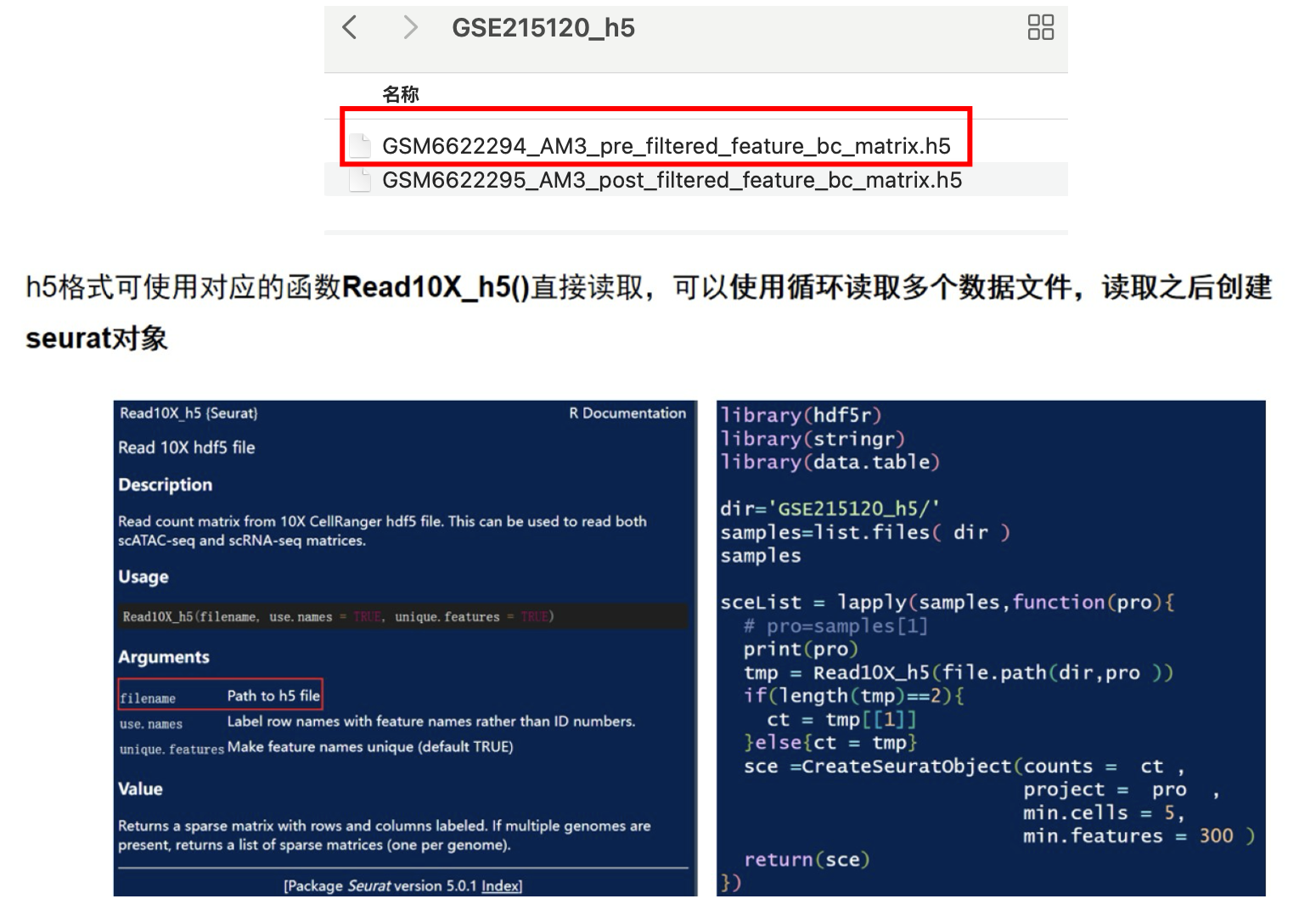
##h5格式
#清空环境 加载需要的R包
rm(list=ls())
options(stringsAsFactors = F)
source('./lib.R')
library(hdf5r)
library(stringr)
library(data.table)
dir='GSE215120_h5/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X_h5(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident) 3 RDS文件
RDS同RData一样,是R语言中存储数据的一种形式,区别于RData直接load就可以,Rds是需要load后赋值的。
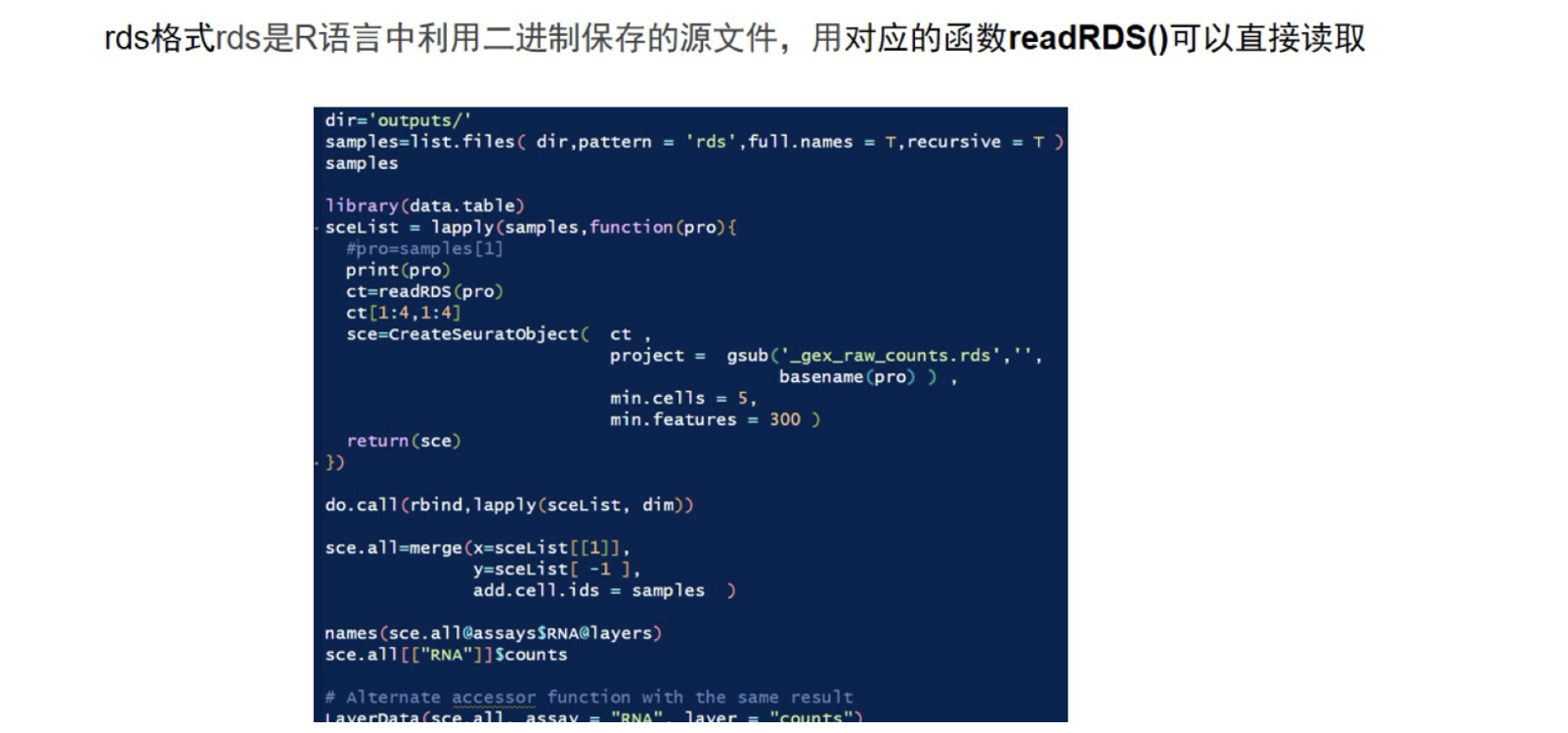
#RDS文件
rm(list=ls())
options(stringsAsFactors = F)
source('./lib.R')
dir='GSE234933_rds/'
samples=list.files( dir,pattern = 'rds',full.names = T,recursive = T )
samples
library(data.table)
sceList = lapply(samples,function(pro){
#pro=samples[1]
print(pro)
ct=readRDS(pro)
ct[1:4,1:4]
sce=CreateSeuratObject( ct ,
project = gsub('_gex_raw_counts.rds','',
basename(pro) ) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)4 txt.gz格式
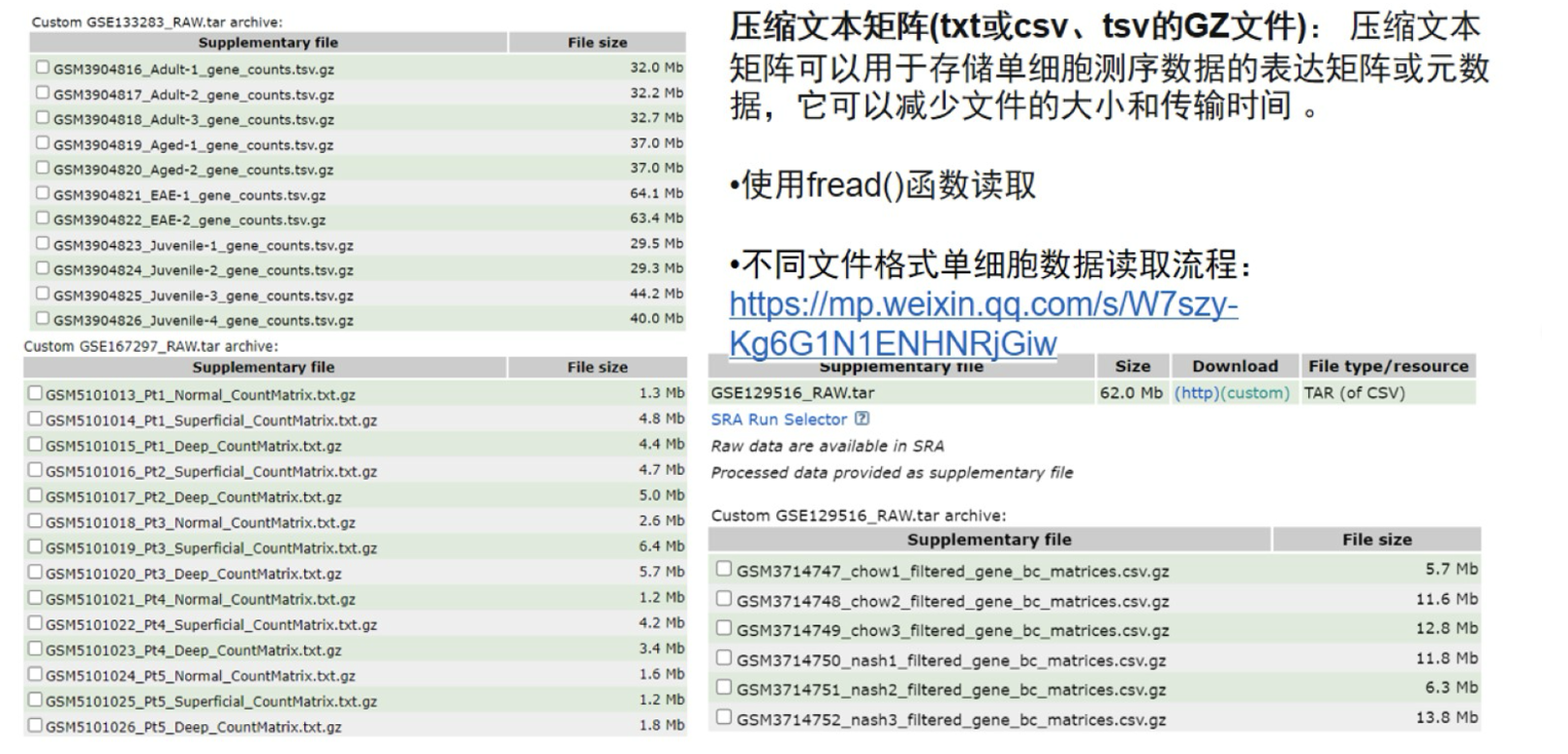
##txt.gz格式
#清空环境 加载需要的R包
rm(list=ls())
options(stringsAsFactors = F)
source('./lib.R')
dir='GSE167297_txt/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_CountMatrix.txt.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident) 5 csv.gz格式
##csv.gz格式
#清空环境 加载需要的R包
rm(list=ls())
options(stringsAsFactors = F)
source('./lib.R')
dir='GSE129516_csv/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_filtered_gene_bc_matrices.csv.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident) 原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号