单细胞空间转录组RCTD去卷积分析学习和整理
原创

RCTD(Robust Cell Type Decomposition),是一种用于将单细胞RNA测序数据中的细胞类型注释转移到空间转录组学数据上的方法。RCTD 通过整合单细胞和空间转录组学数据,能够较为精确地为空间点(spots)分配细胞类型或细胞类型的混合,以便更好地理解空间组织结构中的基因表达情况。
RCTD提供三种模式:
- Doublet mode: 该模式为每个测序点分配1-2种细胞类型,推荐用于具有高空间分辨率的技术,如Slide-seq和MERFISH;
- Full mode: 该模式为每个测序点分配任意数量的细胞类型,推荐用于空间分辨率较低的技术,如100微米分辨率的Visium;
- Multi mode: 这是doublet mode的扩展版本,能够在每个测序点发现超过两种细胞类型,是全模式的替代选项。
每一种都有不同的应用场景,官网提示大多数都可以用Doublet mode
分析步骤
1.导入
rm(list = ls())
library(spacexr)
library(Matrix)
library(Seurat)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 8, progressbar = TRUE))
# 10x Visium
sce_s <- Load10X_Spatial(data.dir = "./RawData/GSE6716963/",
filename = "GSM6716963_19G081_filtered_feature_bc_matrix.h5")
# reference
sc_dataset <- qread("sc_dataset.qs")2.数据预处理
# 过滤
sce_s <- subset(sce_s, nCount_Spatial > 5000)
sce_s <- subset(sce_s, nCount_Spatial < 35000)
# 归一化/找高变/ScaleData
sce_s <- NormalizeData(sce_s)
sce_s <- FindVariableFeatures(sce_s)
sce_s <- ScaleData(sce_s)
# PCA/UMAP
sce_s <- RunPCA(sce_s)
sce_s <- RunUMAP(sce_s, reduction = "pca", dims = 1:50)
# FindNeihbors/Cluster
sce_s <- FindNeighbors(sce_s, reduction = "pca", dims=1:50)
sce_s <- FindClusters(sce_s, resolution = 0.5, verbose = FALSE)
Idents(sce_s) <- sce_s$Spatial_snn_res.0.5
all.markers <- FindAllMarkers(sce_s, only.pos = T)
head(all.markers)
# reference处理##################
# reference细胞数量少一点,多了就好慢
ref <- subset(sc_dataset,downsample = 2000)
ref <- SCTransform(sc_dataset,ncells = 3000,
verbose = FALSE) %>%
RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
# 建议大家都不要用特殊符号
# R语言不喜欢这些特殊符号
a <- ref@meta.data
a$celltype <- sub("/","_",a$celltype)
ref@meta.data <- a
DimPlot(ref, group.by = "celltype", label = TRUE)3.RCTD分析
input数据准备
# 如果抽样之后的细胞数太少(25个?)需要去除
ref <- subset(ref,celltype !="C3"&celltype !="C5")
# extract information to pass to the RCTD Reference function
counts <- ref[["RNA"]]$counts # 官网使用counts
cluster <- as.factor(ref$celltype)
cluster <- droplevels(cluster) # 如果去除了某些celltype
names(cluster) <- colnames(ref)
nUMI <- ref$nCount_RNA
names(nUMI) <- colnames(ref)
reference <- Reference(counts, cluster, nUMI)
# set up query with the RCTD function SpatialRNA
counts <- sce_s[["Spatial"]]@counts
coords <- GetTissueCoordinates(sce_s)
colnames(coords) <- c("x", "y")
coords[is.na(colnames(coords))] <- NULL
query <- SpatialRNA(coords, counts, colSums(counts))RCTD-deconvolution
RCTD <- create.RCTD(query, reference, max_cores = 8)
RCTD <- run.RCTD(RCTD, doublet_mode = "doublet")
qsave(RCTD,"RCTD.qs")
sce_s <- AddMetaData(sce_s, metadata = RCTD@results$results_df)
# 通常关注这个SPOT丰度最高
p1 <- SpatialDimPlot(sce_s, group.by = "first_type")
# SPOT丰度第二高
p2 <- SpatialDimPlot(sce_s, group.by = "second_type")
p1+p2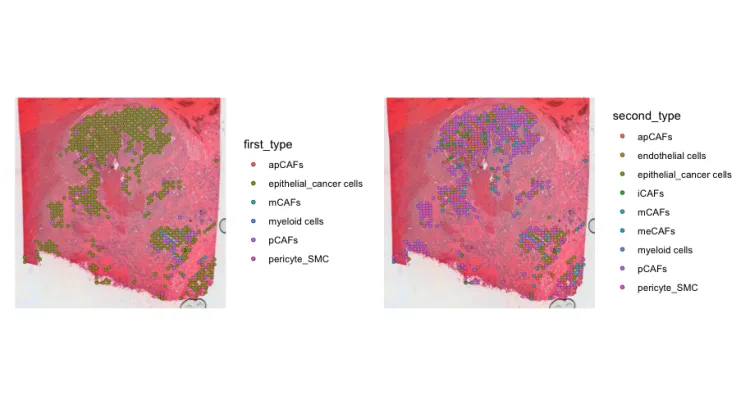
4.RCTD结果可视化
barcodes <- colnames(RCTD@spatialRNA@counts)
weights <- RCTD@results$weights
norm_weights <- normalize_weights(weights)
# observe weight values
celltypes <- c('pericyte_SMC', 'endothelial cells','mast cells')
print(head(norm_weights[,celltypes]))
head(norm_weights)
# plot
p <- plot_puck_continuous(RCTD@spatialRNA,
barcodes,
norm_weights[,'pericyte_SMC'],
ylimit=c(0,0.5),
title='plotofDentateweights',
size=4.5,alpha=0.8);p
ggsave("Spaital_weights.pdf",width=8,height=6,plot=p,bg="white")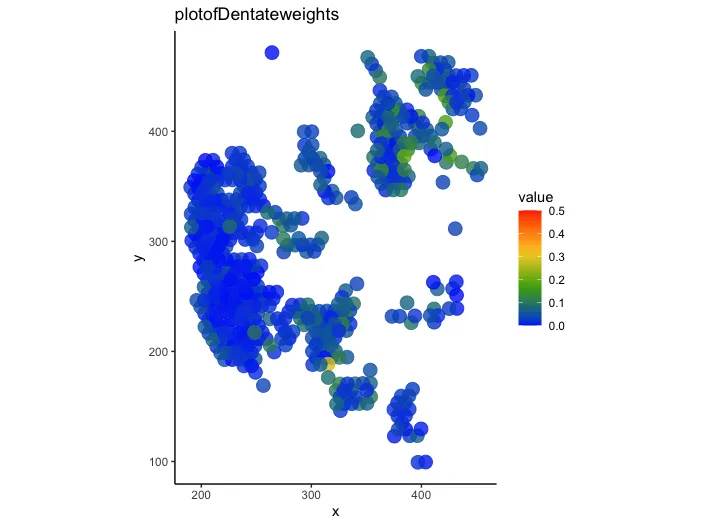
STdeconvolve绘制
library(STdeconvolve)
library(ggplot2)
library(ggsci)
packageVersion("STdeconvolve")
m <- as.matrix(norm_weights)
p <- coords
plt <- vizAllTopics(theta = m,
pos = p,
topicOrder=seq(ncol(m)),
topicCols=rainbow(ncol(m)),
groups = NA,
group_cols = NA,
r = 3, # size of scatterpies; adjust depending on the coordinates of the pixels
lwd = 0.3,
showLegend = TRUE,
plotTitle = "scatterpies")
## function returns a `ggplot2` object, so other aesthetics can be added on:
plt <- plt + ggplot2::guides(fill=ggplot2::guide_legend(ncol=2));plt
ggsave("Spaital_scatterpies.pdf", width=9, height=6, plot=plt, bg="white")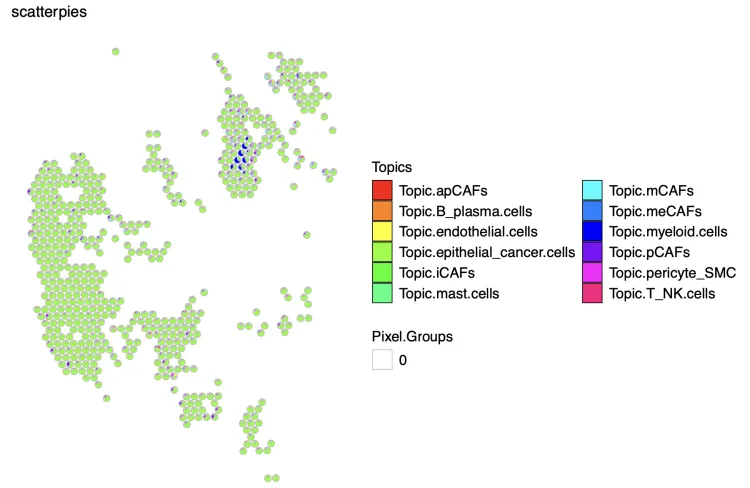
参考资料:
- Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2022 Apr;40(4):517-526
- RCTD-Seirat:https://satijalab.org/seurat/articles/spatial_vignette.html#working-with-multiple-slices-in-seurat
- github:https://github.com/dmcable/spacexr/tree/master/vignettes
- 单细胞天地:https://mp.weixin.qq.com/s/r78K4tm44VqV2FsIoS71Cg
- 朴素的科研打工仔:https://mp.weixin.qq.com/s/jmNw8yxqV3IsMeLAIJpubg
- 单细胞空间交响乐:https://mp.weixin.qq.com/s/y8gfRA91IaHt-svFKdhuHA
单细胞空转分析流程
既往推文:
单细胞空间转录组分析流程学习(一):https://mp.weixin.qq.com/s/E4WuPokBOxKRbBF6CEB5aA
单细胞空间转录组分析流程学习(二): https://mp.weixin.qq.com/s/8AFeq50Dc91cI_6jdQZfFg
单细胞空间转录组分析流程学习python版(三): https://mp.weixin.qq.com/s/wt4nVi8pTFwQMNN8Ipj_mA
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号