TCGA数据库| 如何将表达矩阵与样本临床数据进行合并?


前面我们已经给大家介绍过TCGA数据库中样本barcode的详细组成:TCGA样本barcode详细介绍,现在我们来看看如何将基因表达矩阵与样本临床信息进行合并,方便后续做 比如生存分析,基因在不同样本分期、性别、年龄分组等中的差异表达情况。
首先我们去TGCA下载如乳腺癌的基因表达矩阵
这里使用R包 TCGAbiolinks 去TCGA官网下载数据。
1、加载包:
## download tcga data
## update: 2024-02-22
## Author: zhang juan
rm(list=ls())
# 当然,需要先去安装这个包,如果已安装就可以跳过:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("TCGAbiolinks")
## load packages
library(TCGAbiolinks)
library(SummarizedExperiment)
suppressPackageStartupMessages(library(tidyverse))
2、癌症类型选择:
# 癌症类型,用 getGDCprojects()$project_id 查看所有
getGDCprojects()$project_id
# [1] "TARGET-AML" "MATCH-Z1I" "HCMI-CMDC" "MATCH-W"
# [5] "MATCH-Z1D" "MATCH-Z1A" "MATCH-U" "MATCH-Q"
# [9] "TCGA-PCPG" "MATCH-H" "MATCH-C1" "TCGA-THYM"
# [13] "MATCH-I" "MATCH-S1" "MATCH-P" "MATCH-R"
# [17] "MATCH-Z1B" "TCGA-PAAD" "TCGA-STAD" "TCGA-TGCT"
# [21] "MATCH-S2" "TCGA-SARC" "TCGA-PRAD" "TCGA-READ"
# [25] "TCGA-UCS" "TCGA-UVM" "TRIO-CRU" "VAREPOP-APOLLO"
# [29] "WCDT-MCRPC" "TARGET-ALL-P1" "REBC-THYR" "TARGET-ALL-P2"
# ...
不同缩写代表的含义可取这个地址查看:https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/bcr-batch-codes
本次乳腺癌缩写为:BRCA

3、下载:
# 设置query
query <- GDCquery(
project = "TCGA-BRCA", # 癌症类型,用 getGDCprojects()$project_id 查看所有
data.category="Transcriptome Profiling", # 数据类别, 用getProjectSummary(project)查看所有类别
data.type ="Gene Expression Quantification", # 数据类型
workflow.type="STAR - Counts" # 工作流类型
)
## 下载数据
GDCdownload(query=query, files.per.chunk= 50, directory = "./")
下来后的数据为一个样本一个tsv文件:如 8d1641ea-7552-4d23-9298-094e0056386a.rna_seq.augmented_star_gene_counts.tsv
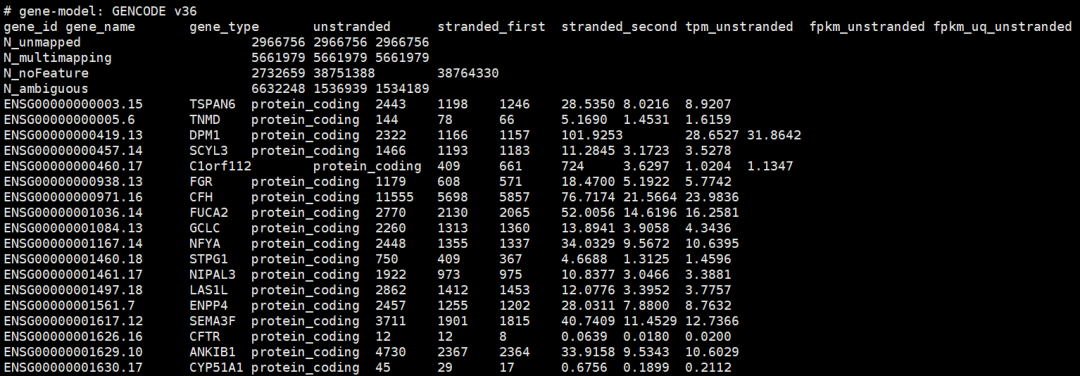
4、整合成一个表达矩阵:
## 整理数据并存储为 R对象
GDCprepare(query,save=T,save.filename="TCGA-BRCA.transcriptome.Rdata", directory = "./")
## 再次加载
load("TCGA-BRCA.transcriptome.Rdata")
ls()
names(assays(data))
rowdata <- rowData(data)
5、提取mRNA的SummarizedExperiment对象,根据gene_type取子集,太简单了!
table(rowdata$gene_type)
tcga_mrna <- data[rowdata$gene_type == "protein_coding",]
tcga_mrna_count <- assay(tcga_mrna,"unstranded") # mRNA的counts矩阵
tcga_mrna_tpm <- assay(tcga_mrna, "tpm_unstrand") # mRNA的tpm矩阵
tcga_mrna_fpkm <- assay(tcga_mrna,"fpkm_unstrand") # mRNA的fpkm矩阵
# 添加gene_symbol, 先提取gene_name
symbol_mrna <- rowData(tcga_mrna)$gene_name
head(symbol_mrna)
####################################################### count值
tcga_mrna_count_symbol <- cbind(data.frame(symbol_mrna), as.data.frame(tcga_mrna_count))
# 去重复保留最大的那个
tcga_mrna_count_symbol1 <- tcga_mrna_count_symbol %>%
as_tibble() %>% # tibble不支持row name,我竟然才发现!
mutate(meanrow = rowMeans(.[,-1]), .before=2) %>%
arrange(desc(meanrow)) %>%
distinct(symbol_mrna,.keep_all=T) %>%
select(-meanrow)
saveRDS(tcga_mrna_count_symbol1, file = "tcga_mrna_count_symbol.rds")
write.table(tcga_mrna_count_symbol1, file ="tcga_mrna_count_symbol.xls",row.names = F,sep = "\t",quote = F)
####################################################### tpm值
tcga_mrna_tpm_symbol <- cbind(data.frame(symbol_mrna), as.data.frame(tcga_mrna_tpm))
# 去重复保留最大的那个
tcga_mrna_tpm_symbol1 <- tcga_mrna_tpm_symbol %>%
as_tibble() %>% # tibble不支持row name,我竟然才发现!
mutate(meanrow = rowMeans(.[,-1]), .before=2) %>%
arrange(desc(meanrow)) %>%
distinct(symbol_mrna,.keep_all=T) %>%
select(-meanrow)
saveRDS(tcga_mrna_tpm_symbol1, file = "tcga_mrna_tpm_symbol.rds")
write.table(tcga_mrna_tpm_symbol1, file = "tcga_mrna_tpm_symbol.xls",row.names = F,sep = "\t",quote = F)
####################################################### fpkm值
tcga_mrna_fpkm_symbol <- cbind(data.frame(symbol_mrna), as.data.frame(tcga_mrna_fpkm))
# 去重复保留最大的那个
tcga_mrna_fpkm_symbol1 <- tcga_mrna_fpkm_symbol %>%
as_tibble() %>% # tibble不支持row name,我竟然才发现!
mutate(meanrow = rowMeans(.[,-1]), .before=2) %>%
arrange(desc(meanrow)) %>%
distinct(symbol_mrna,.keep_all=T) %>%
select(-meanrow)
saveRDS(tcga_mrna_fpkm_symbol1, file = "tcga_mrna_fpkm_symbol.rds")
write.table(tcga_mrna_fpkm_symbol1, file = "tcga_mrna_fpkm_symbol.xls",row.names = F,sep = "\t",quote = F)
接着下载样本临床信息
1、同样首先需要联网 进行 query:
##############################################################################
########################## 3.批量下载临床数据 ###################################
##############################################################################
# ref: https://bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/clinical.html
query <- GDCquery(
project = "TCGA-BRCA",
data.category = "Clinical",
data.format = "bcr xml"
)
save(query, file = "TCGA-BRCA.clinic.query.rdata")
# 下载到当前目录
GDCdownload(query, files.per.chunk= 50, directory = "./")
2、对下载的数据进行整理:
clinical <- GDCprepare_clinic(query, clinical.info = "patient", directory = "./")
clinical.follow_up <- GDCprepare_clinic(query, clinical.info = "follow_up", directory = "./")
clinical.stage_event <- GDCprepare_clinic(query, clinical.info = "stage_event", directory = "./")
clinical.drug <- GDCprepare_clinic(query, clinical.info = "drug", directory = "./")
clinical.radiation <- GDCprepare_clinic(query, clinical.info = "radiation", directory = "./")
clinical.admin <- GDCprepare_clinic(query, clinical.info = "admin", directory = "./")
# 保存
saveRDS(clinical, file = "TCGA-BRCA.clinical_patient.rds")
saveRDS(clinical.admin, file = "TCGA-BRCA.clinical_admin.rds")
saveRDS(clinical.drug, file = "TCGA-BRCA.clinical_drug.rds")
saveRDS(clinical.follow_up, file = "TCGA-BRCA.clinical_follow_up.rds")
saveRDS(clinical.radiation, file = "TCGA-BRCA.clinical_radiation.rds")
saveRDS(clinical.stage_event, file = "TCGA-BRCA.clinical_stage_event.rds")
现在将基因表达矩阵与临床信息整合在一起
先看看各自的样本ID名,根据前面的介绍《TCGA样本barcode详细介绍》,可以看到 表达矩阵里面的是样本ID,临床信息中是patient ID,一个病人可能会取多个样本,比如同时存在正常样本与肿瘤样本,也可能同时具有好几个肿瘤样本:
# 表达矩阵 样本名
mrna_fpkm <- readRDS("tcga_mrna_fpkm_symbol.rds")
head(colnames(mrna_fpkm))
# [1] "symbol_mrna" "TCGA-5L-AAT0-01A-12R-A41B-07" "TCGA-A2-A04U-01A-11R-A115-07" "TCGA-AN-A04A-01A-21R-A034-07"
# [5] "TCGA-A7-A13D-01A-13R-A12P-07" "TCGA-BH-A201-01A-11R-A14M-07"
# 临床信息
clinical <- readRDS(file = "TCGA-BRCA.clinical_patient.rds")
colnames(clinical)
head(clinical[,1:6])
# 我们后面相比较不同病理分期间某个基因表达差异,这里过滤一下样本
clinical <- clinical[,c("bcr_patient_barcode", "stage_event_pathologic_stage")]
colnames(clinical) <- c("bcr_patient_barcode", "pathologic_stage")
str(clinical)
table(clinical$pathologic_stage)
clinical$pathologic_stage <- as.character(clinical$pathologic_stage)
clinical <- clinical[clinical$pathologic_stage!="",]
clinical <- na.omit(clinical)
head(clinical)
# 变成 stage I 、II、III、IV、
clinical$stage <- clinical$pathologic_stage
clinical$stage[grepl("Stage I$|Stage IA$|Stage IB$",clinical$pathologic_stage)] <- "Stage I"
clinical$stage[grepl("Stage II$|Stage IIA$|Stage IIB$",clinical$pathologic_stage)] <- "Stage II"
clinical$stage[grepl("Stage III$|Stage IIIA$|Stage IIIB$|Stage IIIC$",clinical$pathologic_stage)] <- "Stage III"
table(clinical$stage)
table(clinical$pathologic_stage,clinical$stage)
clinical$stage <- factor(clinical$stage, levels = c("Stage I","Stage II","Stage III","Stage IV"))

那么,这里对应的时候,一般可以先将样本分为肿瘤样本与正常样本,看看肿瘤样本中 某个基因表达的高低分组 生存曲线KM差异:
肿瘤样本的编号一般为样本barcode中的第14-15位编码字符:

01-09为肿瘤样本,10以及10以上的为对照样本。肿瘤样本里面又有很多细小的分类:
https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/sample-type-codes
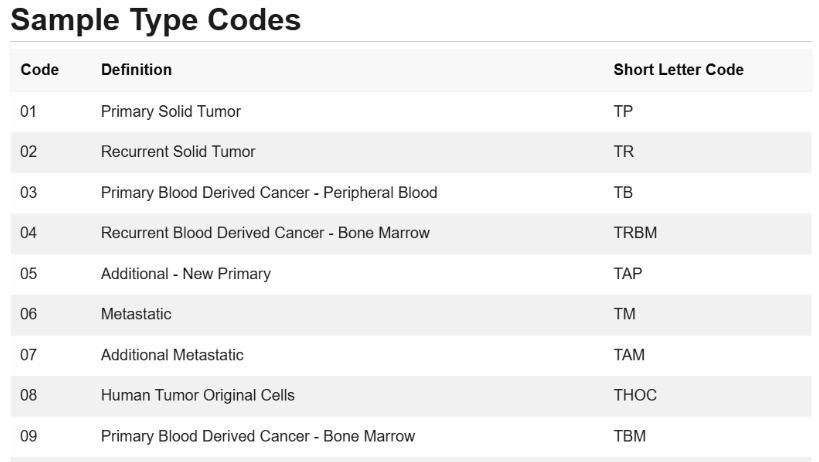
我们这里直接提取 01A类的实体瘤样本:
# 提取 01A类的实体瘤样本
table(str_sub(colnames(mrna_fpkm),14,16))
mrna_fpkm_tumor <- as.data.frame(mrna_fpkm[, str_sub(colnames(mrna_fpkm),14,16)=="01A"])
rownames(mrna_fpkm_tumor) <- mrna_fpkm$symbol_mrna
mrna_fpkm_tumor[1:6,1:6]
# 截取样本名字前面12个字符,与临床信息中的样本ID保持一致
colnames(mrna_fpkm_tumor) <- str_sub(colnames(mrna_fpkm_tumor), 1,12)
head(colnames(mrna_fpkm_tumor))
#[1] "TCGA-5L-AAT0" "TCGA-A2-A04U" "TCGA-AN-A04A" "TCGA-A7-A13D" "TCGA-BH-A201" "TCGA-BH-A0H6"
具有临床信息的病人ID与肿瘤样本表达矩阵取交集:
clinical_com <- clinical[match(comid, clinical$bcr_patient_barcode) ,]
mrna_fpkm_tumor_com <- mrna_fpkm_tumor[, comid]
dim(clinical_com)
# [1] 1056 114
dim(mrna_fpkm_tumor_com)
# [1] 19938 1056
查看乳腺癌中的明星基因 BRCA1 基因在不同分组中的差异吧:
# 查看 brca1基因在不同分组中的差异吧
data <- data.frame(clinical_com, BRCA1=t(mrna_fpkm_tumor_com["BRCA1",]))
head(data)
刚好使用我们前面给大家介绍的绘图小技巧《带有疾病进展的多分组差异结果如何展示?》中的代码绘制:
# 绘制小提琴图和显著性标记
library(ggplot2)
library(ggstatsplot)
library(patchwork)
library(reshape2)
library(stringr)
library(ggsignif)
library(ggsci)
max_pos <- max(data$BRCA1)
max_pos
B <- ggplot(data=data,aes(x=stage,y=BRCA1,colour = stage)) +
geom_boxplot(mapping=aes(x=stage,y=BRCA1,colour = stage), size=0.6, width = 0.5) + # 箱线图
geom_jitter(mapping=aes(x=stage,y=BRCA1,colour = stage),size=1.2) + # 散点
scale_color_npg() +
# #scale_color_manual(limits=c("Stage I","Stage II","Stage III","Stage IV"),
# values =c( "#ed1a22","#00a651","#652b90","") ) + # 颜色
geom_signif(mapping=aes(x=stage,y=BRCA1), # 不同组别的显著性
comparisons = list(c("Stage I","Stage II"),c("Stage III","Stage IV"),c("Stage I","Stage III")),
map_signif_level=T, # T显示显著性,F显示p value
tip_length=c(0,0,0,0,0,0,0,0,0,0,0,0), # 修改显著性线两端的长短
y_position = c(max_pos,max_pos*1.04,max_pos*1.05), # 设置显著性线的位置高度
size=0.8, # 修改线的粗细
textsize = 4, # 修改显著性标记的大小
test = "t.test") + # 检验的类型,可以更改
theme_classic() + #设置白色背景
labs(x="",y="") + # 添加标题,x轴,y轴标签
ggtitle(label = "BRCA1") +
theme(plot.title = element_text(hjust = 0.5),
axis.line=element_line(linetype=1,color="black",size=0.9),
axis.text.x = element_text(size = 12))
B
结果如下:
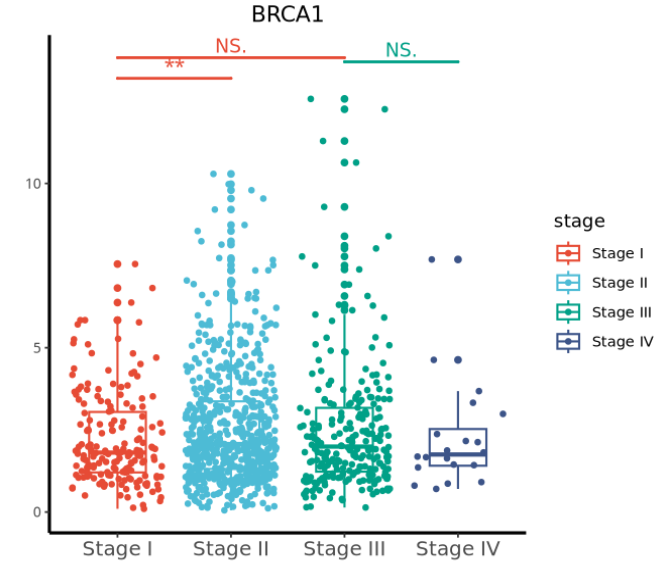
学会了这个,后面就可以随意绘制任意基因在任意临床表型分组间的差异了!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-07,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号