单细胞测序数据分析|手动注释


之前我们分享过单细胞自动注释包GPTtypecell的使用方法。今天我们详细介绍一下细胞类型注释的内容,并且学习手动注释是如何实现的。
YEAR-END SUMMARY
单细胞注释简介
在单细胞转录组分析(scRNA-seq)中,每个细胞都可以测量到大量基因的表达值。为了更好地理解细胞功能和组织组成,需要对每个细胞进行分类并标注其生物学意义,这个过程称为单细胞类型注释。通常,注释是通过结合基因表达数据和先验生物学知识进行的。
单细胞注释的原理主要基于细胞类型特异性基因表达模式。不同类型的细胞会有一组特异性表达的基因(称为标志性基因或Marker基因)。例如:免疫细胞:T细胞通常高表达 CD3D,B细胞高表达 CD19。
上皮细胞:高表达 EPCAM。
成纤维细胞:高表达 COL1A1。
神经元:高表达 MAP2 或 RBFOX3 (NeuN)。
不过,通过标志基因对细胞类型进行分配有一定的局限性,我们知道B细胞会高表达B细胞表面抗原MS4A1,但是我们所测得的B细胞却有着不同的状态,如幼稚B细胞,成熟B细胞,浆细胞等,甚至还有一些疾病/组织特异性B细胞。故细胞类型的注释并不是绝对的,我们一般会首先获得大类细胞,再从大类细胞中识别细胞类型的"亚型"或"细胞状态"(例如,激活状态与静息状态)。
YEAR-END SUMMARY
手动注释
建议从一个稍微复杂的数据开始注释,虽然说简单的数据注释流程与复杂的是一样的,但是太过简单的数据会使得我们无法理解细胞亚群的含义。这里我们选择了降维完成后的人类骨髓数据进行注释。
加载数据:
配置环境:
import omicverse as ov
print(f'omicverse version: {ov.__version__}')
import scanpy as sc
print(f'scanpy version: {sc.__version__}')
ov.ov_plot_set()读取数据
adata = ov.read('s4d8_dimensionality_reduction.h5ad')
adata
聚类
在单细胞测序分析中,先聚类后注释是一个经典流程,而且第一步注释的聚类分辨率不用太高。
sc.pp.neighbors(adata, n_neighbors=15,
n_pcs=30, use_rep='scaled|original|X_pca')
sc.tl.leiden(adata, key_added="leiden_res1", resolution=1.0)
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
ov.utils.embedding(adata,
basis='X_mde',
color=["leiden_res1"],
title=['Clusters'],
palette=ov.palette()[:],
show=False, frameon='small',)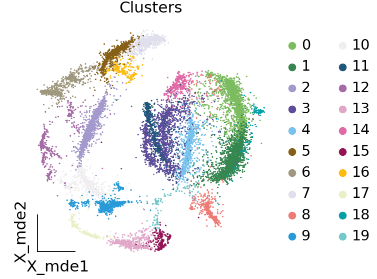
基于marker字典的注释
下面列出了一组基于文献的骨髓细胞标记基因,这些基因由研究特定细胞类型及亚型的论文报告。需要注意,蛋白水平上的标记基因(如用于流式细胞术)在转录组数据中可能表现不佳,因此基于RNA的标记基因通常更可靠。同时,某些标记基因可能在不同数据集中的表现存在差异,理想情况下应在多个数据集中验证其有效性。最后,与领域专家合作尤为重要,生物学家对组织、生物学特性、细胞类型及标记基因更熟悉,有助于提高注释准确性。对于亚群标记基因,应结合多篇文献进行综合分析和验证。
marker_genes = {'Fibroblast':['ACTA2'],
'Endothelium':['PTPRB','PECAM1'],
'Epithelium':['KRT5','KRT14'],
'Mast cell':['KIT','CD63'],
'Neutrophil' :['FCGR3A','ITGAM'],
'cDendritic cell':['FCER1A','CST3'],
'pDendritic cell':['IL3RA','GZMB','SERPINF1','ITM2C'],
'Monocyte' :['CD14','LYZ','S100A8','S100A9','LST1',],
'Macrophage':['CSF1R','CD68'],
'B cell':['MS4A1','CD79A'],
'Plasma cell':['MZB1','IGKC','JCHAIN'],
'Proliferative signal':['MKI67','TOP2A','STMN1'],
'NK/NKT cell':['GNLY','NKG7','KLRD1'],
'T cell':['CD3D','CD3E'],
}在我们的数据中仅选择检测到的标记基因。我们将循环遍历所有marker字典里的细胞类型,并仅保留我们在adata对象中发现的基因作为该细胞类型的标记基因。这将在我们开始绘图时防止错误的发生。
smarker_genes_in_data = dict()for ct, markers in small_marker_dict.items():
markers_found = list() for marker in markers: if marker in adata.var.index:
markers_found.append(marker)
smarker_genes_in_data[ct] = markers_found#del [] # remove the last markerdel_markers = list()for ct, markers in smarker_genes_in_data.items(): if markers == []:
del_markers.append(ct)for ct in del_markers: del smarker_genes_in_data[ct]基于上述字典得到的maker如下:
smarker_genes_in_data
sc.pl.dotplot(
adata,
groupby="leiden_res1",
var_names=smarker_genes_in_data,
dendrogram=True,
standard_scale="var", # standard scale: normalize each gene to range from 0 to 1)
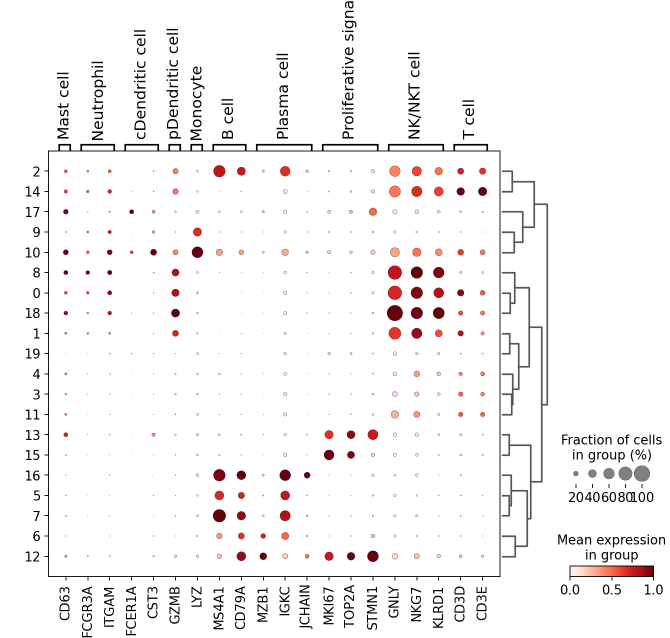
我们可视化这部分细胞,观察注释的细胞群是否连在一起,如果连在一起证明我们注释的正确性,否则注释可能有错误。
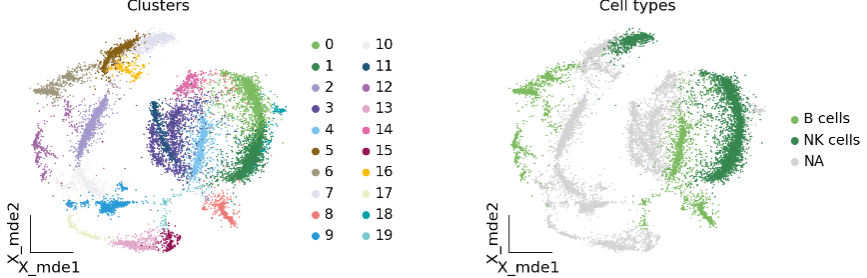
# create a dictionary to map cluster to annotation labelcluster2annotation = { '7': 'NK cells', # Germ-cell(Oid)'1': 'NK cells', # Germ-cell(Oid)'20': 'NK cells', '0': 'NK cells', '19': 'B cells', '4': 'B cells', '8': 'B cells', '6': 'B cells', '12': 'B cells',
}
adata.obs['major_celltype'] = adata.obs['leiden_res1'].map(cluster2annotation).astype('category')
ov.utils.embedding(
adata,
basis='X_mde',
color=['leiden_res1', 'major_celltype'],
title=['Clusters', 'Cell types'],
palette=ov.palette()[:],
wspace=0.55,
show=False,
frameon='small',
)
基于marker数据库的注释
根据上面的分析,我们也可以直接利用数据库来查找特定的细胞类型,我们将使用AUCell来评估每一类细胞的marker基因。
adata_raw = adata.raw.to_adata()
adata_raw
pathway_dict = ov.utils.geneset_prepare(
'CellMarker_Augmented_2021.txt', organism='Human'
)
# Assess all pathways
adata_aucs = ov.single.pathway_aucell_enrichment(
adata_raw,
pathways_dict=pathway_dict,
num_workers=8
)
adata_aucs.obs = adata_raw[adata_aucs.obs.index].obs
adata_aucs.obsm = adata_raw[adata_aucs.obs.index].obsm
adata_aucs.obsp = adata_raw[adata_aucs.obs.index].obsp
adata_aucs首先从Enrichr中下载CellMarker_Augmented_2021数据库,该库一共包含了1074种细胞类型,并且平均每一种细胞类型有80个基因,我们导入该数据库。然后将原来的obs属性赋予新生成的adata_aucs,使其保持原来的细胞元数据。
与此前计算簇特异性基因的过程一样,在这里,同样计算簇特异的细胞类型。
# adata_aucs.uns['log1p']['base'] = None
sc.tl.rank_genes_groups(
adata_aucs, groupby="leiden_res1", use_raw=False,
method="wilcoxon", key_added="dea_leiden_aucs_res1"
)
sc.pl.rank_genes_groups_dotplot(
adata_aucs, groupby='leiden_res1',
cmap='RdBu_r', key='dea_leiden_aucs_res1',
standard_scale='var', n_genes=3
)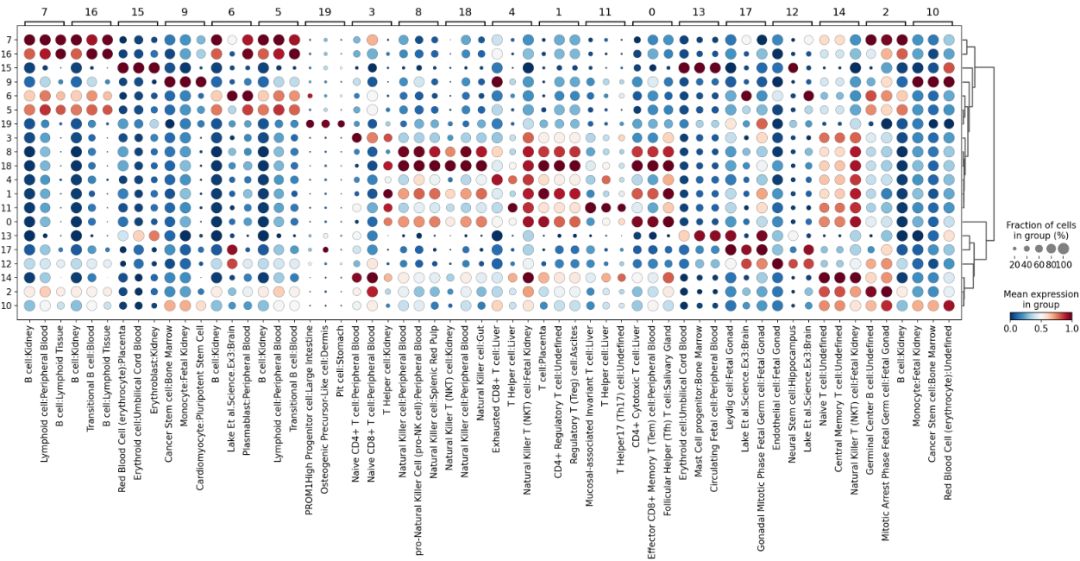
从图中可以发现簇14主要富集了各类的T细胞,我们验证一下每一个簇的细胞类型。
special_cluster = '14'
degs = sc.get.rank_genes_groups_df(
adata_aucs, group=special_cluster, key='dea_leiden_aucs_res1'
)
degs = degs.dropna()
print(
'{}: {}'.format(
special_cluster,
str(
degs.names[:2].tolist()
).replace("'", "")
.replace("[", "")
.replace("]", "")
.replace(",", "|")
)
)
# Loop through clusters
for cluster in adata_aucs.uns['dendrogram_leiden_res1']['categories_ordered']:
special_cluster = str(cluster)
degs = sc.get.rank_genes_groups_df(
adata_aucs, group=special_cluster, key='dea_leiden_aucs_res1'
)
degs = degs.dropna()
print(
'{}: {}'.format(
special_cluster,
str(
degs.names[:2].tolist()
).replace("'", "")
.replace("[", "")
.replace("]", "")
.replace(",", "|")
)
)

需要结合上面的点图,去确定相同的细胞类型的簇
# Create a dictionary to map clusters to annotation labels
cluster2annotation = {
'0': 'NK cells',
'1': 'NK cells',
'2': 'T cells',
'3': 'B cells',
'4': 'B cells',
'5': 'T cells',
'6': 'B cells',
'7': 'NK cells',
'8': 'B cells',
'9': 'Monocyte',
'10': 'T cells',
'11': 'Erythroid',
'12': 'Endothelial cell',
'13': 'Monocyte',
'14': 'T cells',
'15': 'Erythroid',
'16': 'Neurons',
'17': 'Neurons',
'18': 'Erythroid',
'19': 'B cells',
'20': 'NK cells'
}
adata.obs['manual_celltype'] = adata.obs['leiden_res1'].map(cluster2annotation).astype('category')
ov.utils.embedding(
adata,
basis='X_mde',
color=["manual_celltype", "major_celltype"],
title=['Database Cell types', 'Marker Cell types'],
palette=ov.palette()[:],
wspace=0.55,
show=False,
frameon='small'
)
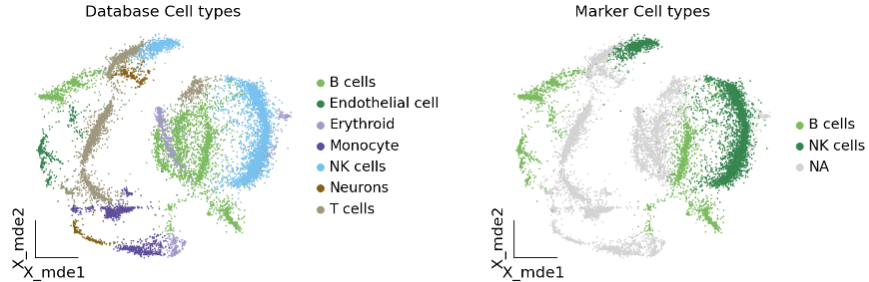
我们发现两种方法所注释得到的细胞高度相似,唯一的区别是我们使用数据库注释的时候会多出一类EndoThelial细胞。我们检查发现这是簇12所注释出来的细胞类型。我们重新研究发现,此前的marker基因的点图中,簇12位于图的尾端,其实其与B细胞不是完全一致,MS4A1的表达也不是特别高。故我们修正该注释结果为Endothelial细胞。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号