综述:ATAC-Seq 数据分析工具大全


技能树今年的新专辑《ATAC-Seq 数据分析2025》会介绍各种关于 ATAC-Seq 数据分析的小知识点,欢迎关注~
今年会在以往的基础上进行迭代与更新,并进行扩展,添加新的内容如scATAC-Seq,欢迎关注新专辑《ATAC-Seq 数据分析2025》~
今天给大家分享的这篇文献综合性地解释了 ATAC-seq 数据处理的基本原理,总结了常见的分析方法,并回顾了计算工具,为不同的研究问题提供建议。这篇文章为 ATAC-seq 数据的分析提供了一个起点和参考。
标题:Analytical Approaches for ATAC-seq Data Analysis 发表:Curr Protoc Hum Genet. 2020 Jun;106(1):e101. doi: 10.1002/cphg.101 链接:https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/cphg.101
ATAC-seq 全称
ATAC-seq 全称为:the Assay for Transpose Accessible Chromatin using sequencing,翻译为转座酶可及染色质测序分析法。
研究目的有:
- 定位核小体
- 识别转录因子结合位点
- 识别对外部因子可及的DNA区域,包括启动子、增强子和其他类型的元件
- 测量DNA调控元件的差异性活性
ATAC-seq的研究数量在短短几年内就接近1万项
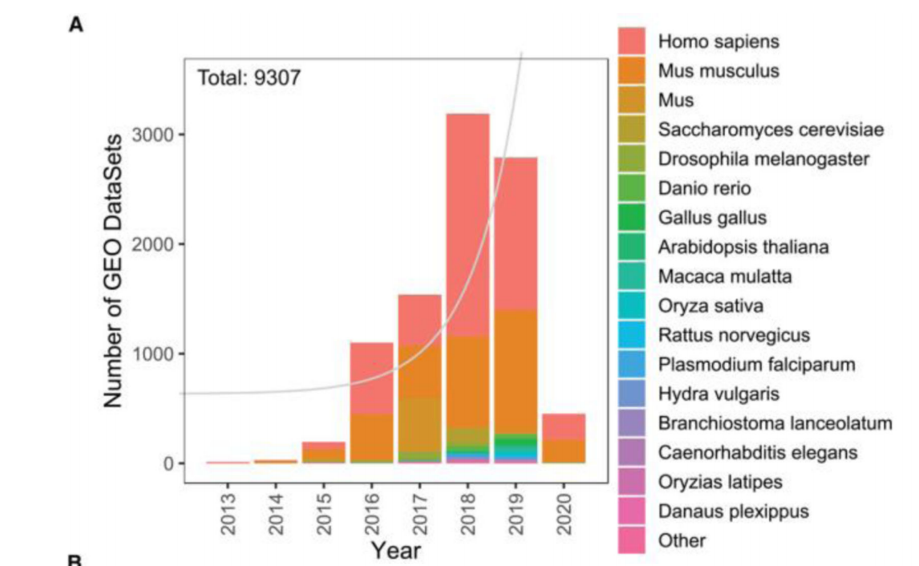
ATAC-seq 实验原理图
ATAC-seq 依赖于一种活跃的Tn5转座酶的活性,Tn5转座酶简介如下:
Tn5转座酶是一种广泛应用于基因组学研究的工具酶,以下是关于Tn5转座酶的详细介绍: 1. 来源与特性 Tn5转座酶来源于大肠杆菌(E. coli),是一种经过改造的突变体,具有极高的活性。它能够特异性识别转座子两端的反向重复序列(如嵌合端Mosaic End, ME),并随机将转座子插入目标DNA序列中。这种转座酶在原核和真核生物的DNA中都表现出高效的插入能力。 2. 作用机制 Tn5转座酶通过形成转座复合体,催化四个磷酸转移反应(包括DNA切割、发夹形成、发夹分解和链转移到目标DNA),从而将转座子整合到新的DNA位点。其插入位点具有一定的随机性,但也有偏好性,首选的DNA靶序列是A-GNT(T/C)(A/T)(A/G)ANC-T。 3. 应用领域 基因组学研究 Tn5转座酶被广泛应用于基因组学研究,尤其是在ATAC-seq(染色质开放性测序)中。它可以识别染色质上的开放区域,剪切DNA片段,并在剪切的同时插入特定序列,从而用于分析基因组的开放性区域。 高通量测序文库构建 Tn5转座酶能够高效地将DNA片段打断并连接接头序列,因此被广泛用于二代测序文库的构建。它能够在单个反应中完成片段化和接头连接,大大简化了文库构建的步骤。 转基因技术 Tn5转座酶可以将外源基因插入宿主细胞基因组中,用于构建转基因细胞系或模型生物。其插入的随机性和高效性使其成为一种理想的基因插入工具。 4. 优势
- 高效性:Tn5转座酶具有极高的活性,能够在短时间内完成DNA片段的插入。
- 随机性:其插入位点具有较高的随机性,适用于需要广泛插入的应用场景。
- 多功能性:除了基因插入,Tn5转座酶还被用于基因组片段化和接头连接,广泛应用于高通量测序。
5. 使用注意事项
- 保存条件:Tn5转座酶通常需要在-80℃保存,解冻后可在-20℃保存2个月。
- 反应体系:在使用Tn5转座酶进行高通量测序文库构建时,需要根据具体应用优化反应体系和条件。
Tn5转座酶因其高效性和多功能性,已成为基因组学研究和高通量测序中不可或缺的工具。
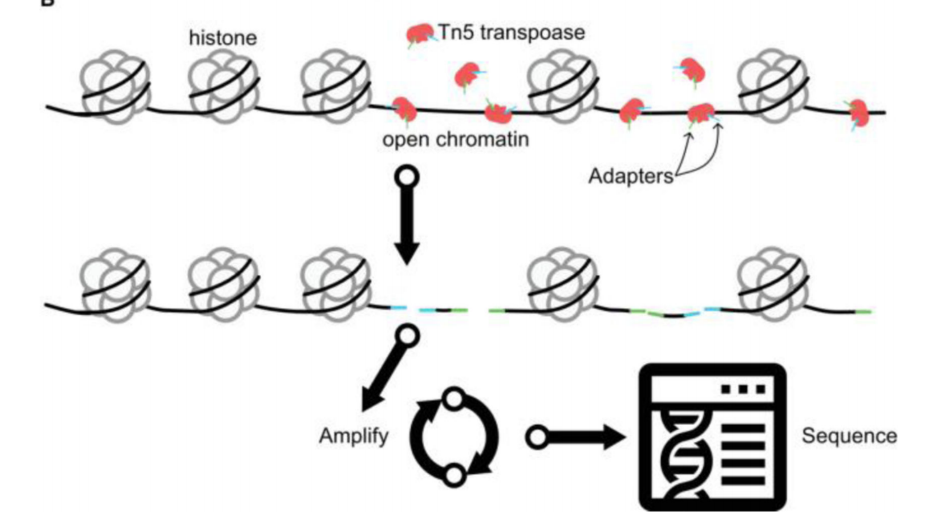
Generalized ATAC-seq library preparation protocol
数据分析流程
- 1、比对, 去接头, 和去除线粒体 reads
- 2、reads 去重复
- 3、生成信号轨迹图
- 4、Peak Calling
- 5、下游分析
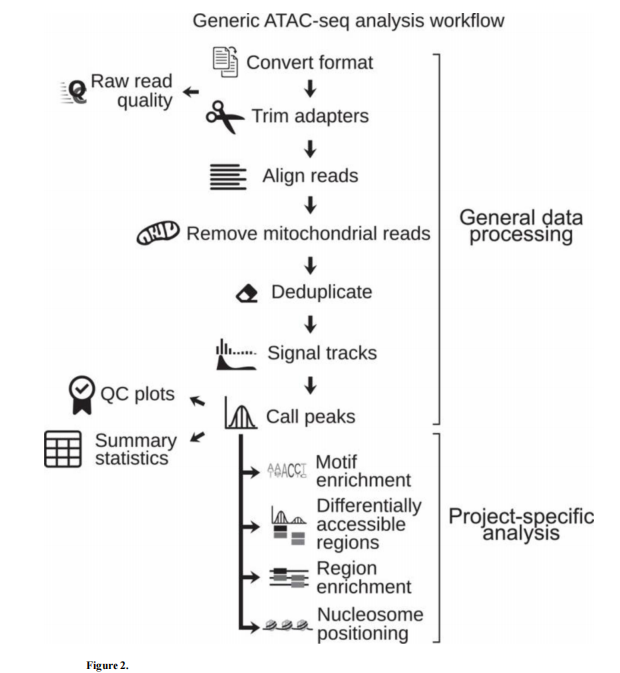
ATAC-seq general workflow
非常详细的 ATAC-seq 数据分析指导资源
Title and | author | Notes | link |
|---|---|---|---|
ATAC-seq data analysis: from FASTQ to peaks | Yiwei Niu,Last updated: 2019 | Blog style walkthrough of generalized ATAC-seq data analysis. | https://yiweiniu.github.io/blog/2019/03/ATAC-seq-data-analysis-from-FASTQ-to-peaks/ |
BIOINF525 Lab 3.2 | Steve Parker,Last updated: 2016 | Minimal standard ATAC-seq analysis walkthrough. | https://github.com/ParkerLab/ |
Analysis of ATAC-seq data in R and Bioconductor | Rockefeller Bioinformatics Resource, Last updated: 2018 | Bioconductor ATAC-seq analysis course. | https://rockefelleruniversity.github.io/RU_ATACseq/ |
ATAC-seq | John M. Gaspar,Last updated: 2019 | Generalized ATAC-seq analysis walkthrough with included custom scripts. | https://github.com/harvardinformatics/ATAC-seq |
ATAC-seq data analysis | Delisle L; Doyle M; & Heyl F,Last updated: 2020 | Galaxy training walkthrough of generalized ATAC-seq analysis. | https://galaxyproject.github.io/training-material/topics/epigenetics/tutorials/atac-seq/tutorial.html |
ATAC-seq 原始数据处理 Pipelines
软件名 | Language | Notes | Docs | Citation |
|---|---|---|---|---|
AIAP | Bash; R; Python | Optimized analysis with novel QC metrics | ++ | Liu et al. (2019) Last updated: 2019 |
ATAC2GRN | Bash; Python | Parameter optimized ATAC-seq pipeline | + | Pranzatelli, Michael, & Chiorini (2018) Last updated: 2018 |
ATAC-pipe | Python; R | Analysis pipeline for ATAC-seq data including TF footprinting; cell-type classification; and regulatory network creation | +++ | Zuo et al. (2019) Last updated: 2019 |
ATACProc | Bash; Python; R | Complete pipeline with additional downstream analyses included | ++ | Unpublished Last updated: 2019 |
Basepair | NA | Commercial. Web-based GUI for complete analysis | ? | Unpublished |
CIPHER | R; Perl; Python | A data processing platform for ChIP-seq; RNA-seq; MNase-seq; DNase-seq; ATAC-seq; and GRO-seq datasets | + | Guzman & D’Orso (2017) Last updated: 2017 |
ENCODE | Python; Bash | Complete pipeline following ENCODE standards for ATAC/DNase-seq analysis | ++ | Unpublished Last updated: 2020 |
esATAC | R | Complete pipeline including downstream analyses | +++ | Wei, Zhang, Fang, Li, & Wang (2018) Last updated: 2019 |
GUAVA | Java; Python; R | GUI based complete ATAC-seq pipeline | + | Divate & Cheung (2018) Last updated: 2019 |
I-ATAC | Java | GUI based interactive ATAC-seq pipeline | + | Ahmed & Ucar (2017) Last updated: 2017 |
nfcore/atacseq | Python; R | Complete pipeline build using Nextflow | +++ | Ewels et al. (2019) Last updated: 2019 |
PEPATAC | Python; R; Perl | Complete pipeline with unique analytical approaches and QC metrics | +++ | Unpublished Last updated: 2019 |
pyflow-ATACseq | Bash; Python | ATAC-seq snakemake pipeline with included nucleosome positioning and TF footprinting | ++ | Unpublished Last updated: 2019 |
snakePipes ATAC-seq | Python | Workflow system including ATAC-seq analysis | +++ | Bhardwaj et al. (2019) Last updated: 2019 |
Tobias Rausch | Bash; R; Python | Complete pipeline with emphasis on downstream analyses | ++ | Rausch et al. (2019) Last updated: 2020 |
ATAC-seq 数据质控工具
Languages | Notes | Docs | Citation | |
|---|---|---|---|---|
ATAqC | Bash; Python | Generate ATAC-seq specific quality control metrics. | + | Unpublished Last updated: 2017 |
ATACseqQC | R | Provides ATAC-seq specific quality control metrics and transcription factor footprinting. | +++ | Ou et al. (2018) Last updated: 2018 |
ataqv | C++; Bash | ATAC-seq QC and visualization. | +++ | Orchard, Kyono, Hensley, Kitzman, & Parker (2020) Last updated: 2020 |
Peak Calling 工具
软件名 | Languages | Notes | Docs | Citation |
|---|---|---|---|---|
F-Seq | Java | Can be used as general peak caller to identify regions of open chromatin. | ++ | Boyle et al. (2008) Last updated: 2016 |
Genrich | C | Peak caller for genomic enrichment assays with specific ATAC-seq mode. | +++ | unpublished Last updated: 2020 |
HMMRATAC | Java | Identify nucleosome positioning and leverage ATAC-seq specific read outs to call peaks. | +++ | Tarbell & Liu (2019) Last updated: 2020 |
Hotspot2 | C++ | Identify significantly enriched genomic regions. | ++ | Unpublished Last updated: 2019 |
HOMER | Perl; C++ | Suite of tools that include the ability to call peaks from DNA enrichment assays. | +++ | Heinz et al. (2010) Last updated: 2010 |
MACS2 | Python | Specifically designed for CHiP-seq but broadly applicable to any DNA enrichment assay to call peaks. | +++ | Zhang et al. (2020) Last updated: 2020 |
PeaKDEck | Perl | Peak calling program for DNase-seq data. | +++ | McCarthy & O’Callaghan (2014) Last updated: 2014 |
差异可及区域分析工具
软件 | Languages | Notes | Docs | Citation |
|---|---|---|---|---|
DAStk | Python | Identifies changes in transcription factor activity by looking at changes in chromatin accessibility | +++ | Tripodi et al. (2018) Last updated: 2020 |
diffTF | Python; R | Identifies differential transcription factors. Can operate in basic mode with just chromatin accessibility or in classification mode where it integrates RNA-seq. | +++ | Berest et al. (2019) Last updated: 2020 |
Motif 富集 和转录因子 Footprinting 工具
Languages | Notes | Docs | Citation | |
|---|---|---|---|---|
BiFET | R | Identify overrepresented transcription factor footprints. | ++ | Youn et al. (2019) Last updated: 2019 |
BinDNase | R | Transcription factor binding prediction using DNase-seq. | + | Kähärä & Lähdesmäki (2015) Last updated: 2015 |
CENTIPEDE | R | Transcription factor footprinting and binding site prediction. | ++ | Pique-Regi et al. (2011) Last updated: 2010 |
DeFCoM | Python | Detecting transcription factor footprints and underlying motifs using supervised learning. | +++ | Quach & Furey (2017) Last updated: 2017 |
DNase2TF | R | Identify footprint candidates from DNase-seq data on user-specified regions. | + | Sung et al. (2014) Last updated: 2017 |
HINT-ATAC | Python | Use open chromatin data to identify transcription factor footprints with modifications specific to ATAC-seq data. | +++ | Li et al. (2019) Last updated: 2019 |
HOMER | Perl; C++ | A suite of tools for motif discovery and enrichment. | +++ | Heinz et al. (2010) Last updated: 2019 |
MEME Suite | Perl; Python | Suite of tools for motif discovery; enrichment; and GO term analyses. | +++ | Bailey et al. (2009) Last updated: 2020 |
PIQ | Bash; R | Models genome-wide DNase profiles to identify transcription factor binding sites. | ++ | Sherwood et al. (2014) Last updated: 2016 |
TOBIAS | Python | Identify transcription factor footprints. | ++ | Bentsen et al. (2019) Last updated: 2020 |
TRACE | Python | Transcription factor footprinting. | ++ | Ouyang & Boyle (2019) Last updated: 2020 |
Wellington | Python | Identify TF footprints using DNase-seq data. | +++ | Piper et al. (2013) Last updated: 2019 |
核小体定位分析工具
软件 | Languages | Notes | Docs | Citation |
|---|---|---|---|---|
HMMRATAC | Java | Identify nucleosome positioning and leverage ATAC-seq specific read outs to call peaks. | +++ | Tarbell & Liu (2019) Last updated: 2020 |
NucleoATAC | Python; R | Call nucleosomes using ATAC-seq data. | +++ | Schep et al. (2015) Last updated: 2019 |
NucTools | Perl; R | Calculate nucleosome occupancy profiles on chromatin accessibility data. | +++ | Vainshtein et al. (2017) Last updated: 2019 |
区域富集分析工具
软件 | Languages | Notes | Docs | Citation |
|---|---|---|---|---|
Annotatr | R | Annotate summarize and visualize genomic regions. | +++ | Cavalcante & Sartor (2017) Last updated: 2019 |
BART/BARTweb | Python | Predict factors that bind at cis-regulatory regions. | +++ | Wang et al. (2018) Last updated: 2020 |
chipenrich | R | Perform gene set enrichment testing using genomic regions. | +++ | Welch et al. (2014) Last updated: 2020 |
coloc-stats | Python | Perform co-localization analysis of genomic regions. | +++ | Simovski et al. (2018) Last updated: 2019 |
COLO | JSP | Identify genomic features in close proximity to user-submitted genomic regions. | ++ | Kim et al. (2015) Last updated: 2015 |
FEATnotator | Perl; R | Annotate genomic regions. | ++ | Podicheti & Mockaitis (2015) Last updated: 2018 |
GenomeRunner | .NET | Perform annotation and enrichment of genomic regions against default or custom regulatory regions. | ++ | Dozmorov et al. (2016) Last updated: 2016 |
GenometriCorr | R | Determine spatial correlation between region sets. | ++ | Favorov et al. (2012) Last updated: 2020 |
Genomic Association Tester | Python | Calculate the significance of overlaps between multiple genomic region sets. | +++ | Heger et al. (2013) Last updated: 2019 |
GIGGLE | C | Genomics search engine to uncover significantly shared genomic loci (regions) between data. | +++ | Layer et al. (2018) Last updated: 2019 |
GLANET | Java; Perl | Genomic loci annotation and enrichment tool between sets of genomic regions. | +++ | Otlu et al. (2017) Last updated: 2019 |
GREAT | C | Annotate genomic regions. | +++ | McLean et al. (2010) Last updated: 2019 |
LOLA/LOLAweb | R | Determine significant enrichment between region sets to inform on biological meaning. | +++ | Sheffield & Bock (2016) Last updated: 2019 |
regioneR | R | Evaluate significant associations between region sets using permutation testing. | +++ | Gel et al. (2016) Last updated: 2020 |
StereoGene | C++; R | Estimate genome-wide correlation between pairs of genomic features. | ++ | Stavrovskaya et al. (2017) Last updated: 2019 |
单细胞 scATAC-seq 数据处理工具
软件 | Languages | Notes | Docs | Citation |
|---|---|---|---|---|
BAP | R; Python | Bead-based scATAC-seq data processing. | ++ | Lareau et al. (2019) Last updated: 2019 |
BROCKMAN | R; Bash; Ruby | Convert genomics data into K-mer words associated with chromatin marks used to compare and identify changes across samples. | ++ | de Boer & Regev (2018) Last updated: 2018 |
Cell Ranger ATAC | NA | Commercial. Set of analysis pipelines for Chromium single cell ATAC-seq. | +++ | Unpublished |
chromVAR | R | Identify transcription factor accessibility in single-cell data. Enables clustering of single-cell ATAC-seq data. | +++ | Schep et al. (2017) Last updated: 2019 |
Cicero | R | Predict cis-regulatory DNA interactions using single-cell chromatin accessibility data. | +++ | Pliner et al. (2018) Last updated: 2019 |
cisTopic | R | Identify cell states and cis-regulatory topics from single-cell data. | +++ | Bravo González-Blas et al.(2019) Last updated: 2019 |
scABC | R | Classify single-cell ATAC using unsupervised clustering and identify chromatin regions specific to cell identity. | + | Zamanighomi et al. (2018) Last updated: 2019 |
SCALE | Python | Clustering and visualization of single-cell ATAC-seq data into interpretable cell populations. | ++ | Xiong et al. (2019) Last updated: 2019 |
Scasat | Bash; Python; R | Complete pipeline to process scATAC-seq data with simple steps. | +++ | Baker et al. (2019) Last updated: 2019 |
scATAC-pro | R; Python | Comprehensive pipeline for single cell ATAC-seq analysis. | +++ | Yu et al. (2019) Last updated: 2020 |
scOpen | Python | Chromatin-accessibility estimation of single-cell ATAC data. | + | Li et al. (2019) Last updated: 2020 |
SCRAT | R | Useful for studying single cell heterogeneity. Can identify changes in gene sets or transcription factor binding sites. Includes GUI and web-based service. | +++ | Ji et al. (2017) Last updated: 2018 |
SnapATAC | R; Python | Single Nucleus Analysis Pipeline for ATAC-seq. | +++ | Fang et al. (2019) Last updated: 2019 |
此外:作者维护了一个不断扩大的 ATAC-seq 工具列表,可前往关注:
https://github.com/databio/awesome-atac-analysis
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-23,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号