pdx模型的人和小鼠单细胞数据分析方案


什么是pdx模型
首先,有些人比如我可能还不是很了解什么是pdx模型,让人工智能大模型帮我介绍一下。问kimi(https://kimi.moonshot.cn/):什么是pdx模型?
PDX 模型全称为病人来源肿瘤异种移植(Patient-derived tumor xenograft)模型,是将来源于患者的肿瘤组织或原代细胞植入免疫缺陷小鼠体内形成的移植瘤模型。 PDX模型的特点
- 保留肿瘤特性:PDX模型在组织病理学、分子生物学和基因水平上保留了大部分原代肿瘤的特点,包括肿瘤的异质性、关键基因突变以及肿瘤微环境。
- 模拟真实肿瘤:它能够较好地模拟人体肿瘤的生长、转移、血管生成和免疫抑制等过程。
- 临床相关性高:PDX模型的临床疗效预测性较好,能够更真实地反映患者的肿瘤特征和对药物的反应。
PDX模型的应用
- 新药研发:用于药物筛选、药效验证以及临床试验患者的筛选。
- 个性化治疗:帮助识别药物敏感或耐药的生物标志物,为患者提供个性化的治疗方案。
- 肿瘤生物学研究:用于研究肿瘤的异质性、肿瘤干细胞以及肿瘤的进展和演变。
PDX模型的构建方法
- 样本采集:从患者手术或活检中获取新鲜肿瘤组织。
- 移植:将肿瘤组织移植到免疫缺陷小鼠体内,常用的移植方式包括皮下移植和原位移植。
- 传代:待肿瘤在小鼠体内生长到一定大小后,取出并移植到新的小鼠体内,形成多代移植瘤。
PDX模型的局限性
- 物种差异:小鼠与人类存在生物学差异,可能导致实验结果与人体实际反应不完全一致。
- 实验复杂性:构建和维护PDX模型需要较高的技术要求和成本。
- 样本限制:PDX模型缺少配对的正常样本,且从模型中提取的DNA片段可能包含小鼠基因。
PDX模型因其高度的临床相关性和对肿瘤异质性的保留,在癌症研究和治疗中具有重要价值,但也需要克服一些技术挑战。
也就是说,它将来自患者的癌细胞或组织移植到小鼠等动物宿主体内培养。这种模型的目的是更好地模拟人体内癌症的特性,以进行更真实、更有效的药物测试和研究。因此,取样时主要涉及的是人体内的肿瘤组织,但是同时也会有小鼠细胞混合,如果拿去做单细胞转录组建库测序,得到的测序数据里面就会有人、小鼠两个物种。
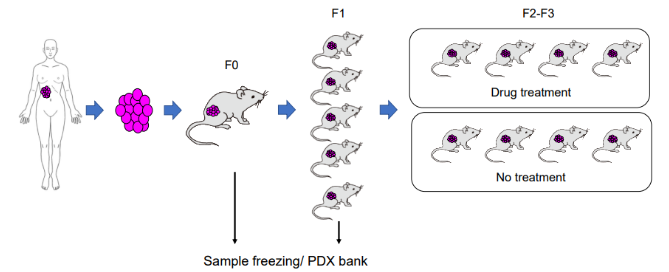
处理方法
问一下人工智能
pdx模型的单细胞转录组如何分析上游定量,参考基因怎么选择呢?
在PDX模型的单细胞转录组分析中,上游定量和参考基因选择是关键步骤,以下是相关分析方法和建议: 1. 单细胞转录组上游定量分析 对于PDX模型的单细胞转录组数据,由于涉及人类和小鼠两种物种的基因表达,需要特别处理以区分人源和鼠源细胞,并准确定量基因表达。以下是分析流程: 数据比对与物种识别
- 将单细胞转录组测序数据与人和小鼠的混合基因组库进行比对,获得基于混合基因组库的细胞-基因表达谱矩阵。
- 根据细胞中表达人基因或小鼠基因的比例,识别细胞为人细胞、小鼠细胞或双细胞。例如,设定一个预设阈值(如70%),当细胞中表达某物种基因的比例大于等于该阈值时,将其归为该物种的细胞。
数据提取与校正
- 基于细胞的barcode,从测序数据中提取识别为人细胞或小鼠细胞的序列。
- 对于部分匹配的序列,如果测序质量值较低(如<10),可以进行校正后再提取。
基因表达谱构建
- 将提取的人细胞序列与人类参考基因组比对,小鼠细胞序列与小鼠参考基因组比对,分别获得人细胞和小鼠细胞的基因表达谱。
2. 参考基因的选择 参考基因的选择对于单细胞转录组分析至关重要,尤其是在PDX模型中,需要同时考虑人类和小鼠的基因。 物种特异性参考基因
- 人类参考基因:选择广泛表达且稳定的基因,如
ACTB(β-actin)、GAPDH(甘油醛-3-磷酸脱氢酶)、HPRT1(次黄嘌呤磷酸核糖转移酶)等。 - 小鼠参考基因:可以选择
Actb、Gapdh、Hprt1等基因。
同源基因处理
- 在PDX模型中,人和小鼠的同源基因可能导致比对混淆。可以通过构建整合基因序列集来解决:将人和小鼠的同源基因序列拼接,并在两者之间插入一定数量的N碱基(如80个N碱基),以防止跨物种比对。
数据过滤与校正
- 对比对结果进行过滤,只保留唯一比对到整合基因的reads,以提高定量的准确性。
3. 分析工具与软件
- 使用
Cell Ranger软件进行数据比对和定量,它支持多物种参考基因组的构建和分析。 - 对于下游分析,可以使用
Seurat等R包进行数据处理、降维、聚类和差异分析。
通过以上方法,可以有效处理PDX模型的单细胞转录组数据,准确区分人源和鼠源细胞,并进行可靠的基因表达定量分析。
上面人工智能告诉我们,可能会有两种分析方法:
第一种:使用 人与小鼠的融合的参考基因组进行比对
如 10x genomics官网非常便利的给我们提供了 人与小鼠的融合参考基因组(refdata-gex-GRCh38_and_GRCm39-2024-A.tar.gz),可以前往下载:
# https://www.10xgenomics.com/support/software/cell-ranger/downloads#reference-downloads
#wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz"
#wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz"
wget -c "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38_and_GRCm39-2024-A.tar.gz"
第二种:分别使用人与小鼠的参考基因进行比对定量
如 2020年5月发表在 Genome Medicine 上的文献《Single-cell RNA sequencing reveals the tumor microenvironment and facilitates strategic choices to circumvent treatment failure in a chemorefractory bladder cancer patient》中,分析策略就为分别对 人和小鼠进行比对,然后采用一个阈值方式来区分两个物种来源的细胞:

2023年10月发表在Cell Rep上的文献《Collagen 1-mediated CXCL1 secretion in tumor cells activates fibroblasts to promote radioresistance of esophageal cancer》中,则只是分析了人源的细胞。

我现在对 第一种比较感兴趣,还没有做过。他跟第二种会有什么不一样呢?
使用来自 文献 Collagen 1-mediated CXCL1 secretion in tumor cells activates fibroblasts to promote radioresistance of esophageal cancer中的数据,下载地址为https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA016013,下载方式见文章:PDX小鼠模型的单细胞样品定量能选择人类参考基因组吗。
然后走cellranger的流程,拿到标准矩阵:10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元),拿到的结果,其中一个样本:
├── CRR727434
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
然后我们可以看一下 features.tsv文件,里面会有两种来源的基因,并用物种的前缀进行了区分:
# 人的基因,GRCh38开头,ENSG00000243485这种ID也可以区分出来
zless -S features.tsv.gz |head
GRCh38_ENSG00000243485 GRCh38_MIR1302-2HG Gene Expression
GRCh38_ENSG00000237613 GRCh38_FAM138A Gene Expression
GRCh38_ENSG00000186092 GRCh38_OR4F5 Gene Expression
GRCh38_ENSG00000238009 GRCh38_AL627309.1 Gene Expression
GRCh38_ENSG00000239945 GRCh38_AL627309.3 Gene Expression
GRCh38_ENSG00000239906 GRCh38_AL627309.2 Gene Expression
GRCh38_ENSG00000241860 GRCh38_AL627309.5 Gene Expression
GRCh38_ENSG00000241599 GRCh38_AL627309.4 Gene Expression
GRCh38_ENSG00000286448 GRCh38_AP006222.2 Gene Expression
GRCh38_ENSG00000236601 GRCh38_AL732372.1 Gene Expression
# 小鼠的基因,mm10开头,ENSMUSG00000096550这种ID带有小鼠物种MUS信息
zless -S features.tsv.gz |tail
mm10___ENSMUSG00000096550 mm10___Gm16367 Gene Expression
mm10___ENSMUSG00000094172 mm10___AC163611.1 Gene Expression
mm10___ENSMUSG00000094887 mm10___AC163611.2 Gene Expression
mm10___ENSMUSG00000091585 mm10___AC140365.1 Gene Expression
mm10___ENSMUSG00000095763 mm10___AC124606.2 Gene Expression
mm10___ENSMUSG00000095523 mm10___AC124606.1 Gene Expression
mm10___ENSMUSG00000095475 mm10___AC133095.2 Gene Expression
mm10___ENSMUSG00000094855 mm10___AC133095.1 Gene Expression
mm10___ENSMUSG00000095019 mm10___AC234645.1 Gene Expression
mm10___ENSMUSG00000095041 mm10___AC149090.1 Gene Expression
读取数据
###
### Create: Jianming Zeng
### Date: 2023-12-31
### Email: jmzeng1314@163.com
### Blog: http://www.bio-info-trainee.com/
### Forum: http://www.biotrainee.com/thread-1376-1-1.html
### CAFS/SUSTC/Eli Lilly/University of Macau
### Update Log: 2023-12-31 First version
### Update Log: 2024-12-09 by juan zhang (492482942@qq.com)
###
rm(list=ls())
options(stringsAsFactors = F)
library(ggsci)
library(dplyr)
library(future)
library(Seurat)
library(clustree)
library(cowplot)
library(data.table)
library(ggplot2)
library(patchwork)
library(stringr)
library(qs)
library(Matrix)
getwd()
# 读取数据
dir='../inputs/'
samples=list.files( dir )
samples
[1] "CRR727434" "CRR727435" "CRR727436" "CRR727437" "CRR727438" "CRR727439"
sceList <- lapply(samples,function(pro){
# pro=samples[1]
print(pro)
counts <- Read10X(file.path(dir,pro ))
sce <- CreateSeuratObject(counts = counts , project = pro, min.cells = 5, min.features = 500 )
print(dim(sce))
return(sce)
})
names(sceList) <- samples
sceList[[1]]
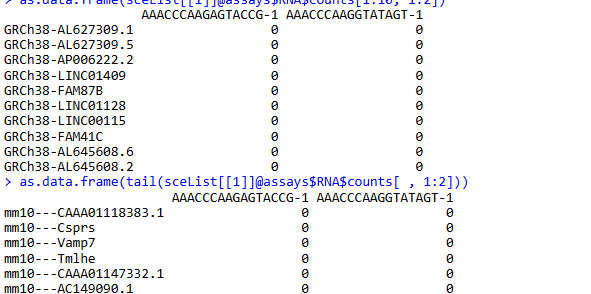
as.data.frame(sceList[[1]]@assays$RNA$counts[1:10, 1:2])
as.data.frame(tail(sceList[[1]]@assays$RNA$counts[ , 1:2]))
可以看到数据中存在两种来源的基因:
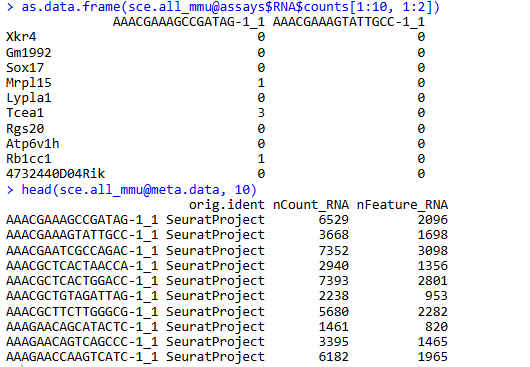
提取小鼠来源的数据
## 提取出来小鼠来源的数据
sceList_mmu <- lapply(sceList,function(sce){
#sce = sceList[[1]]
ct = sce@assays$RNA$counts
kp=grepl('mm10---',rownames(ct)); table(kp)
ct = ct[kp,]
# 去掉前缀
rownames(ct) = gsub('mm10---','',rownames(ct))
ct[1:4,1:4]
sce = CreateSeuratObject( counts = ct, min.cells = 5, min.features = 500)
print(dim(sce))
return(sce)
})
sce.all_mmu <- merge(x=sceList_mmu[[1]], y=sceList_mmu[-1])
sce.all_mmu <- JoinLayers(sce.all_mmu) # seurat v5
sce.all_mmu
as.data.frame(sce.all_mmu@assays$RNA$counts[1:10, 1:2])
head(sce.all_mmu@meta.data, 10)
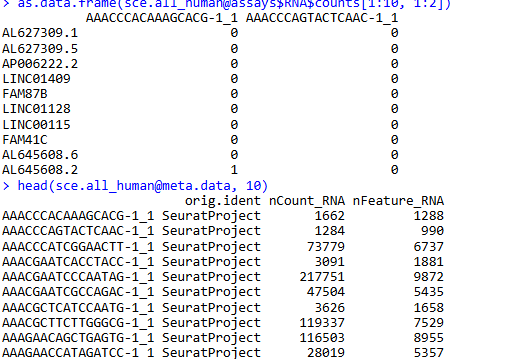
同理,我们也可以很快拿到人来源的数据
################################################################
## 提取出来人来源的数据
sceList_human <- lapply(sceList,function(sce){
#sce = sceList[[1]]
ct = sce@assays$RNA$counts
kp=grepl('GRCh38-',rownames(ct)); table(kp)
ct = ct[kp,]
# 去掉前缀
rownames(ct) = gsub('GRCh38-','',rownames(ct))
ct[1:4,1:4]
sce = CreateSeuratObject( counts = ct, min.cells = 5, min.features = 500)
print(dim(sce))
return(sce)
})
sce.all_human <- merge(x=sceList_human[[1]], y=sceList_human[-1])
sce.all_human <- JoinLayers(sce.all_human) # seurat v5
sce.all_human
as.data.frame(sce.all_human@assays$RNA$counts[1:10, 1:2])
head(sce.all_human@meta.data, 10)
这样我们就得到了两个物种来源的单细胞数据,后面可以分别进行降维聚类分群分析。下一期我们来看看这种混合模式的与分开各自参考基因对比分析的有何不一样~
还有一个疑问:这两种来源的细胞既然在同一个模型中,他们之间的相关性分析有研究做吗?
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号