一个优秀的ATAC-seq数据分析资源实战(一)


之前我们给大家介绍了两篇ATAC-Seq数据分析pipeline的优秀综述:综述:ATAC-Seq 数据分析工具大全 和 Omni-ATAC:更新和优化的ATAC-seq协议(NatProtoc),我们今天就来实战介绍!
这次选择介绍的资源如下,这些内容是通过互联网上的多种资源(论坛、论文、研讨会等)整理而成的。作者无法一一列出所有来源,但想要感谢他们分享经验、知识和代码:
网址:https://yiweiniu.github.io/blog/2019/03/ATAC-seq-data-analysis-from-FASTQ-to-peaks/
1.ATAC-seq概览
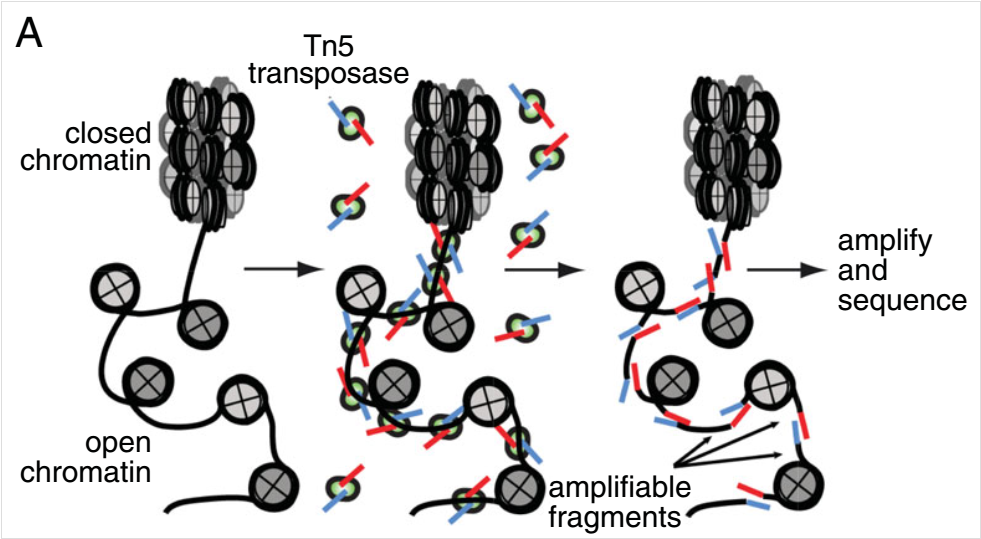
ATAC-seq(转座酶可及染色质高通量测序)是一种用于测定全基因组范围内染色质可及性的方法。它利用一种高活性的Tn5转座酶将测序接头插入到开放染色质区域。图:ATAC-seq overview(Buenrostro et al., 2015).
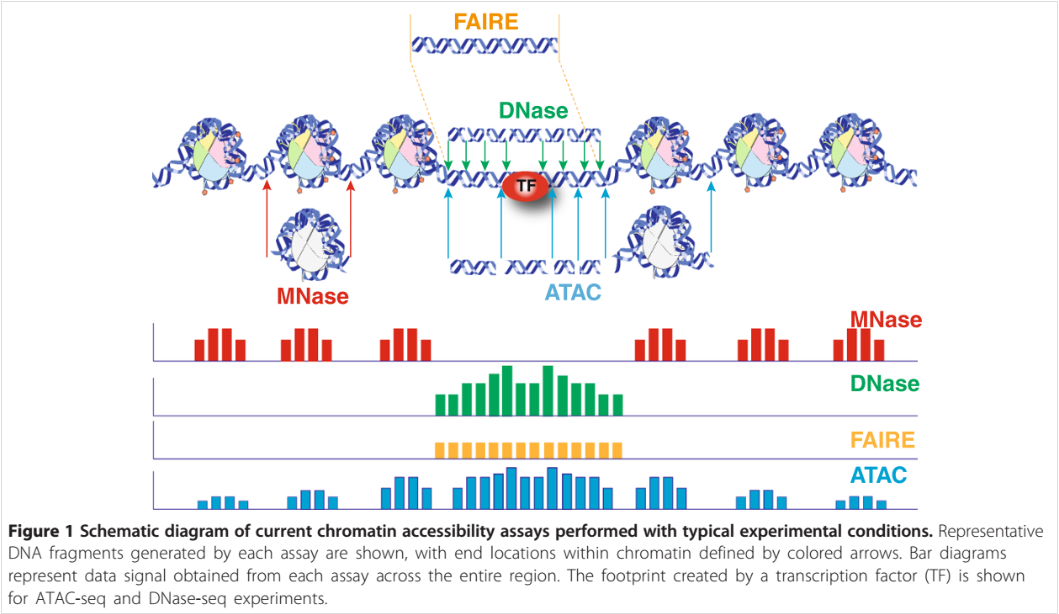
并且这些峰值看起来如下图所示:ATAC-seq peaks (Tsompana and Buck, 2014)
ATAC-Seq可以用来:
- 生成表观基因组学特征
- 在不同组织或条件下绘制可及染色质图谱
- 检索核小体位置
- 识别重要的转录因子
- 生成转录因子的占用特征(足迹分析)
2.实验设计
See Buenrostro et al., 2015, ENCODE - ATAC-seq Data Standards and Prototype Processing Pipeline, and Harvard FAS Informatics - ATAC-seq Guidelines for details.
- 两个或更多的生物学重复样本
- 每个重复样本包含单端测序的2500万条非重复、非线粒体的比对读段,以及双端测序的5000万条
- 通常无需“input”样本
- 在构建文库时,尽可能使用最少的PCR循环次数
- 优先选择双端测序
3.数据分析
Several useful pipelines.
- Harvard FAS Informatics - ATAC-seq Guidelines – clear and up-to-date.
- ENCODE - ATAC-seq Data Standards and Prototype Processing Pipeline
- Tobias Rausch - ATAC-seq analysis pipeline
- ENCODE ATAC-seq pipeline
- Parker Lab - ATAC-seq lab for BIOINF525
- Ferhat Ay Lab - ATAC-seq processing pipeline
- Rockefeller University, ATACseq in R
- 生物信息学生 R 入门教程 - 第五章 ATAC-seq数据分析 – in R.
3.0 数据下载
这里使用数据 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171832 为示例分析数据,里面包含两个样本的ATAC-Seq数据,对应的BipProject编号为:PRJNA721095。从ENA数据库(https://www.ebi.ac.uk/ena/browser/view/PRJNA721095)下载样本的表型信息:filereport_read_run_PRJNA721095_tsv.txt
# 激活小环境
conda activate rna
# 创建项目目录
mkdir 2022-GSE171832-28-embryonal-atacseq
cd 2022-GSE171832-28-embryonal-atacseq
# 创建分析目录
mkdir -p data/rawdata data/cleandata Mapping PeakCalling PeakAnno
cd data/rawdata
# 下载数据
# 项目编号:PRJNA721095
# fq下载 ascp
# 生成down.sh
cat fq.url |while read id
do
echo "ascp -v -QT -l 300m -P33001 -k1 -i /nas2/zhangj/biosoft/miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh era-fasp@${id} ./ "
done >down.sh
# 下载
nohup bash down.sh 1>down.log 2>&1 &
fq.url
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/036/SRR14205836/SRR14205836_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/036/SRR14205836/SRR14205836_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/037/SRR14205837/SRR14205837_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/037/SRR14205837/SRR14205837_2.fastq.gz
down.sh的内容:
ascp -v -QT -l 300m -P33001 -k1 -i /nas2/zhangj/biosoft/miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/036/SRR14205836/SRR14205836_1.fastq.gz ./
ascp -v -QT -l 300m -P33001 -k1 -i /nas2/zhangj/biosoft/miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/036/SRR14205836/SRR14205836_2.fastq.gz ./
ascp -v -QT -l 300m -P33001 -k1 -i /nas2/zhangj/biosoft/miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/037/SRR14205837/SRR14205837_1.fastq.gz ./
ascp -v -QT -l 300m -P33001 -k1 -i /nas2/zhangj/biosoft/miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR142/037/SRR14205837/SRR14205837_2.fastq.gz ./
3.1 数据QC
像分析其他NGS测序数据一样,只有原始FASTQ文件需要进行质量控制,目前有许多可用的软件,例如 Trimmomatic 和 Trim Galore。
首先进行Trim Galore:
# Trim Galore 质控
cd data/cleandata
ls ../rawdata/*1.fastq.gz | awk -F'/' '{print $NF}' |cut -d '_' -f 1 >ID
# 生成trim.sh
cat ID | while read id
do
echo "trim_galore --cores 30 -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ./ ../rawdata/${id}_1.fastq.gz ../rawdata/${id}_2.fastq.gz"
done >trim.sh
# 运行trim.sh, 使用parafly进行并行任务投递
nohup ParaFly -c trim.sh -CPU 2 1>trim.log 2>&1 &
trim.sh的内容如下:
trim_galore --cores 30 -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ./ ../rawdata/SRR14205836_1.fastq.gz ../rawdata/SRR14205836_2.fastq.gz
trim_galore --cores 30 -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ./ ../rawdata/SRR14205837_1.fastq.gz ../rawdata/SRR14205837_2.fastq.gz
接着看一下过滤后的fq质量,看fastqc报告:
multiqc *.zip -n qc_trim
看完后发现,作者上传的是过滤后的cleandata。
3.2 比对和过滤
接下来是比对到参考基因组上,两种流行的比对工具是 BWA 和 Bowtie2。我将使用 Bowtie2(因为它在许多教程和论文中被广泛使用)。
首先还需要下载参考基因组,去 https://asia.ensembl.org/index.html 下载小鼠的fa以及 gtf 和 gff3
cd ~/database/genome/Mus_musculus/release113
wget -c https://ftp.ensembl.org/pub/release-113/fasta/mus_musculus/dna/Mus_musculus.GRCm39.dna.primary_assembly.fa.gz
wget -c https://ftp.ensembl.org/pub/release-113/gtf/mus_musculus/Mus_musculus.GRCm39.113.gtf.gz
wget -c https://ftp.ensembl.org/pub/release-113/gff3/mus_musculus/Mus_musculus.GRCm39.113.gff3.gz
# 下载完整后解压
gunzip *gz
接着构建索引并比对
## 构建索引
bowtie2-build Mus_musculus.GRCm39.dna.primary_assembly.fa Mus_musculus.GRCm39.dna.primary_assembly.fa
## bowtie2比对
cd /nas1/zhangj/project/2022-GSE171832-28-embryonal-atacseq/Mapping
# better alignment results are frequently achieved with --very-sensitive
# use -X 2000 to allow larger fragment size (default is 500)
Bowtie2Index=/nas1/zhangj/database/genome/Mus_musculus/release113/Mus_musculus.GRCm39.dna.primary_assembly.fa
# 生成bowtie2.sh
cat ../data/cleandata/ID | while read id
do
echo "bowtie2 --very-sensitive -X 2000 -x $Bowtie2Index -1 ../data/cleandata/${id}_1_val_1.fq.gz -2 ../data/cleandata/${id}_2_val_2.fq.gz -p 35 2>${id}.bowtie2.log | samtools sort -@ 35 -O bam -o ${id}.sorted.bam -"
done >bowtie2.sh
# 运行bowtie2.sh, 使用parafly进行并行任务投递
nohup ParaFly -c bowtie2.sh -CPU 2 1>bowtie2.log 2>&1 &
# samtools 构建bam索引
samtools index -@ 60 -M SRR14205836.sorted.bam SRR14205837.sorted.bam
bowties.sh 的内容如下:
bowtie2 --very-sensitive -X 2000 -x /nas1/zhangj/database/genome/Mus_musculus/release113/Mus_musculus.GRCm39.dna.primary_assembly.fa -1 ../data/cleandata/SRR14205836_1_val_1.fq.gz -2 ../data/cleandata/SRR14205836_2_val_2.fq.gz -p 35 2>SRR14205836.bowtie2.log | samtools sort -@ 35 -O bam -o SRR14205836.sorted.bam -
bowtie2 --very-sensitive -X 2000 -x /nas1/zhangj/database/genome/Mus_musculus/release113/Mus_musculus.GRCm39.dna.primary_assembly.fa -1 ../data/cleandata/SRR14205837_1_val_1.fq.gz -2 ../data/cleandata/SRR14205837_2_val_2.fq.gz -p 35 2>SRR14205837.bowtie2.log | samtools sort -@ 35 -O bam -o SRR14205837.sorted.bam -
两个样本的总比对率都在95%以上。
Mitochondrial reads
Ref: Harvard FAS Informatics - ATAC-seq Guidelines
由于线粒体基因组中不存在我们感兴趣的ATAC-seq峰值,这些 reads 只会使后续步骤变得复杂。因此,作者建议通过以下方法之一将它们从进一步分析中移除:
- 在比对reads之前,从参考基因组中移除线粒体基因组。在人类/小鼠基因组构建中,线粒体基因组被标记为“chrM”。可以在构建基因组索引之前从参考基因组中删除该序列。这种方法的缺点是比对结果看起来会更糟,因为所有线粒体 reads 都会被计为未比对。
- 在比对后移除线粒体reads。在ATAC-seq模块中提供了一个名为removeChrom的Python脚本来完成这一任务。
由于 mtDNA-reads 的占比是文库质量的一个指标,我们通常在比对后移除线粒体reads。其运行方式如下:
# 生成rmChrM.sh
cat ../data/cleandata/ID | while read id
do
echo "samtools view -@ 35 -h ${id}.sorted.bam | grep -v chrM | samtools sort -@ 35 -O bam -o ${id}.rmChrM.bam -"
done >rmChrM.sh
# 运行rmChrM.sh, 使用parafly进行并行任务投递
nohup ParaFly -c rmChrM.sh -CPU 2 1>rmChrM.log 2>&1 &
# samtools 构建bam索引
samtools index -@ 60 -M SRR14205836.rmChrM.bam SRR14205837.rmChrM.bam
PCR duplicates
Ref: Harvard FAS Informatics - ATAC-seq Guidelines
PCR重复是指在PCR过程中产生的DNA片段的精确拷贝。由于它们是文库制备过程中的产物,可能会干扰我们感兴趣的生物学信号。因此,作为分析流程的一部分,应当将它们移除。 用于移除PCR重复的一个常用程序是Picard的MarkDuplicates工具。
# 安装一下 picard
wget https://github.com/broadinstitute/picard/releases/download/3.3.0/picard.jar
# 测试一下是否可以使用
java -jar picard.jar -h
# 生成 rmDup.sh
# REMOVE_DUPLICATES=false: mark duplicate reads, not remove.
# Change it to true to remove duplicate reads.
cat ../data/cleandata/ID | while read id
do
echo "java -XX:ParallelGCThreads=30 -Djava.io.tmpdir=/tmp -jar /nas2/zhangj/biosoft/picard/picard.jar MarkDuplicates QUIET=true INPUT=${id}.rmChrM.bam OUTPUT=${id}.rmDup.bam METRICS_FILE=${id}.sorted.metrics REMOVE_DUPLICATES=true CREATE_INDEX=true VALIDATION_STRINGENCY=LENIENT TMP_DIR=/tmp "
done >rmDup.sh
# 运行rmDup.sh, 使用parafly进行并行任务投递
nohup ParaFly -c rmDup.sh -CPU 2 1>rmDup.log 2>&1 &
(未完待续~)
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号