Nat. Methods | AF2BIND: 利用AlphaFold2配对表示预测小分子结合位点

Nat. Methods | AF2BIND: 利用AlphaFold2配对表示预测小分子结合位点

DrugAI
发布于 2026-03-30 18:39:38
发布于 2026-03-30 18:39:38
DRUGONE
在蛋白质结构中准确识别小分子结合位点是药物发现的重要任务。然而,真正意义上的从头结合位点预测仍然具有挑战性。研究人员提出AF2BIND模型,该方法利用AlphaFold2内部的配对表示作为特征,并通过逻辑回归模型预测蛋白质中可能与小分子配体接触的残基。
AF2BIND无需依赖同源建模、多序列比对或已知配体信息,即可实现结合位点预测。模型不仅能够预测结合位点位置,还可以通过模型解释分析推测潜在配体的化学性质。研究人员进一步将该方法应用于整个人类蛋白质组,预测出大量此前未被识别的潜在小分子结合位点。该方法为蛋白功能研究和药物靶点发现提供了新的计算工具。

准确识别蛋白质中的配体结合位点是结构生物学和药物设计中的核心问题。传统方法通常依赖同源结构信息,通过结构相似性将已知结合位点转移到新蛋白上。然而,对于缺乏同源模板的蛋白质,这类方法往往难以应用。
另一类方法通过几何或物理特征寻找潜在口袋。例如,一些工具通过在蛋白表面布置探针点或使用α球算法检测可能的结合腔。这些方法在识别明显口袋方面表现良好,但在浅表或非典型结合位点上往往效果有限。近年来,深度学习模型也被用于结合位点预测,但从零训练的模型通常面临训练数据有限和泛化能力不足的问题。
随着大型蛋白质基础模型的发展,研究人员开始探索这些模型所学习到的结构表示是否能够迁移到新的任务。例如,AlphaFold2、ESM等模型在海量蛋白序列和结构数据上进行了预训练,其内部特征可能包含丰富的结构和功能信息。研究人员因此提出一个问题:AlphaFold2在预测蛋白结构过程中所学习到的表示,是否能够用于识别小分子结合位点。
(图2占位)
方法概述
研究人员设计了一种基于AlphaFold2内部表示的结合位点预测方法。首先,将目标蛋白序列和其主链结构输入AlphaFold2模型,同时引入20种不同的“诱饵”氨基酸残基作为独立链加入模型。
AlphaFold2在处理这些输入时会生成配对表示,即描述蛋白残基之间关系的高维特征张量。研究人员提取目标蛋白残基与20个诱饵氨基酸之间的配对特征,并将这些特征输入逻辑回归模型,从而预测每个残基成为小分子结合位点的概率。
模型训练使用来自蛋白质数据库的大量蛋白–小分子复合物结构,并通过严格的数据划分避免训练数据与测试数据之间的相似性,从而确保模型能够评估真正的从头预测能力。
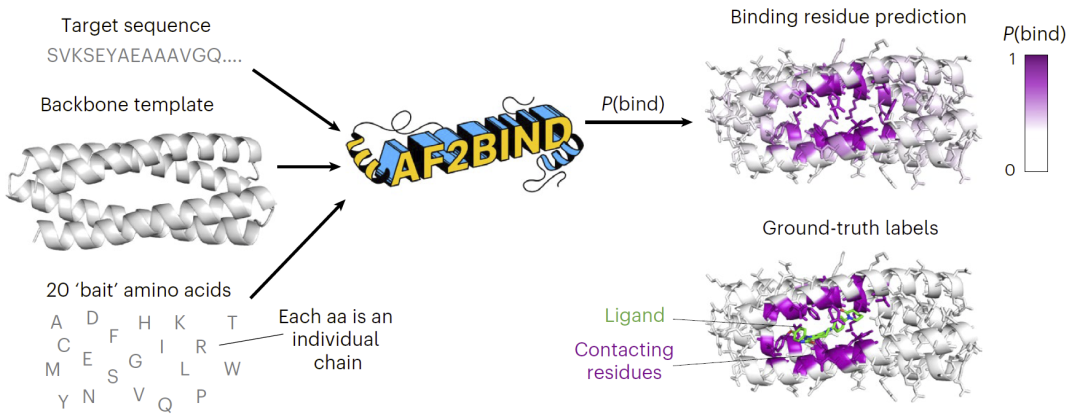
图1|AF2BIND利用AlphaFold2特征预测目标蛋白中的小分子结合残基。
结果
AF2BIND模型框架
AF2BIND通过利用AlphaFold2的配对表示来预测每个残基参与小分子结合的概率。模型输入包括目标蛋白序列、结构模板以及20种诱饵氨基酸。通过分析这些诱饵残基与蛋白残基之间的相互作用信号,模型能够识别潜在结合位点。
由于模型采用逻辑回归结构,其预测结果具有较高可解释性。研究人员能够直接分析不同诱饵氨基酸对预测结果的贡献,从而理解不同氨基酸类型在结合位点识别中的作用。
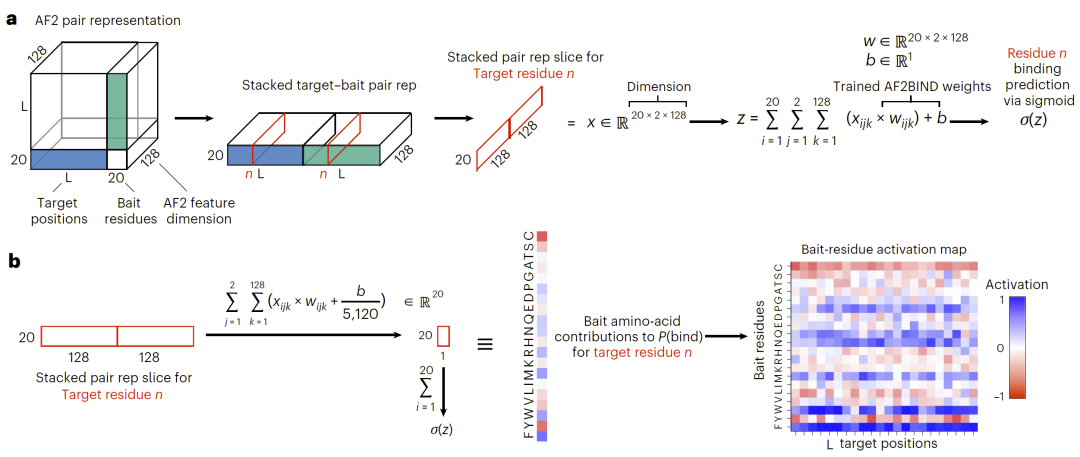
图2|AlphaFold2的配对表示作为输入,用于逻辑回归模型AF2BIND预测配体结合残基。
AlphaFold2配对表示提升预测性能
研究人员比较了不同预训练模型表示在结合位点预测任务中的表现,包括AlphaFold2、ESM2和ESM1-IF等。
结果显示,AlphaFold2的配对表示在单一特征中表现最佳。AF2BIND在测试数据上的结合残基识别率达到约66%,ROC AUC约为0.94,明显优于其他单一表示模型。当与其他模型特征结合时,预测性能还可以进一步提升。
这些结果表明,AlphaFold2在结构预测任务中学习到的内部表示能够有效捕捉与小分子结合相关的结构特征。
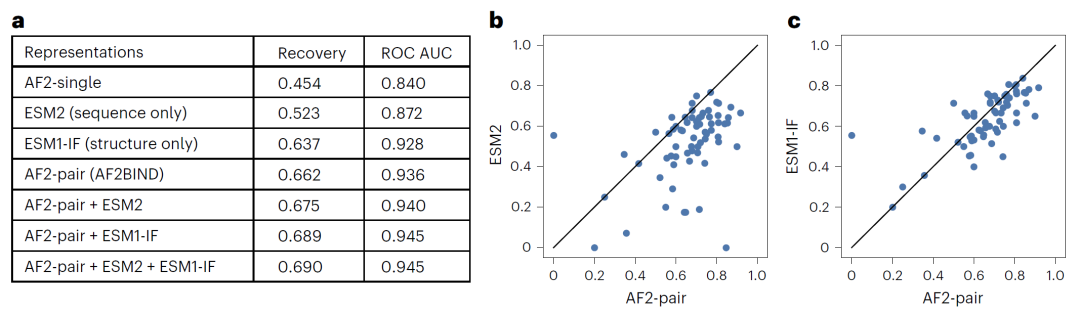
图3|AlphaFold2配对表示在结合残基预测任务中表现最佳
在未见蛋白类别上的预测能力
研究人员进一步测试AF2BIND在未参与训练的蛋白类别上的表现,例如G蛋白偶联受体和溴结构域蛋白。结果表明,即使在完全未见过的蛋白类别上,AF2BIND仍能准确识别结合残基,并给出残基参与结合的概率排序。
模型预测结果对结构变化具有较高鲁棒性。即使蛋白在结合配体前后存在一定构象变化,模型仍能保持较高预测准确度。不过,在某些结合口袋在无配体状态下完全塌陷的情况下,预测性能会有所下降。
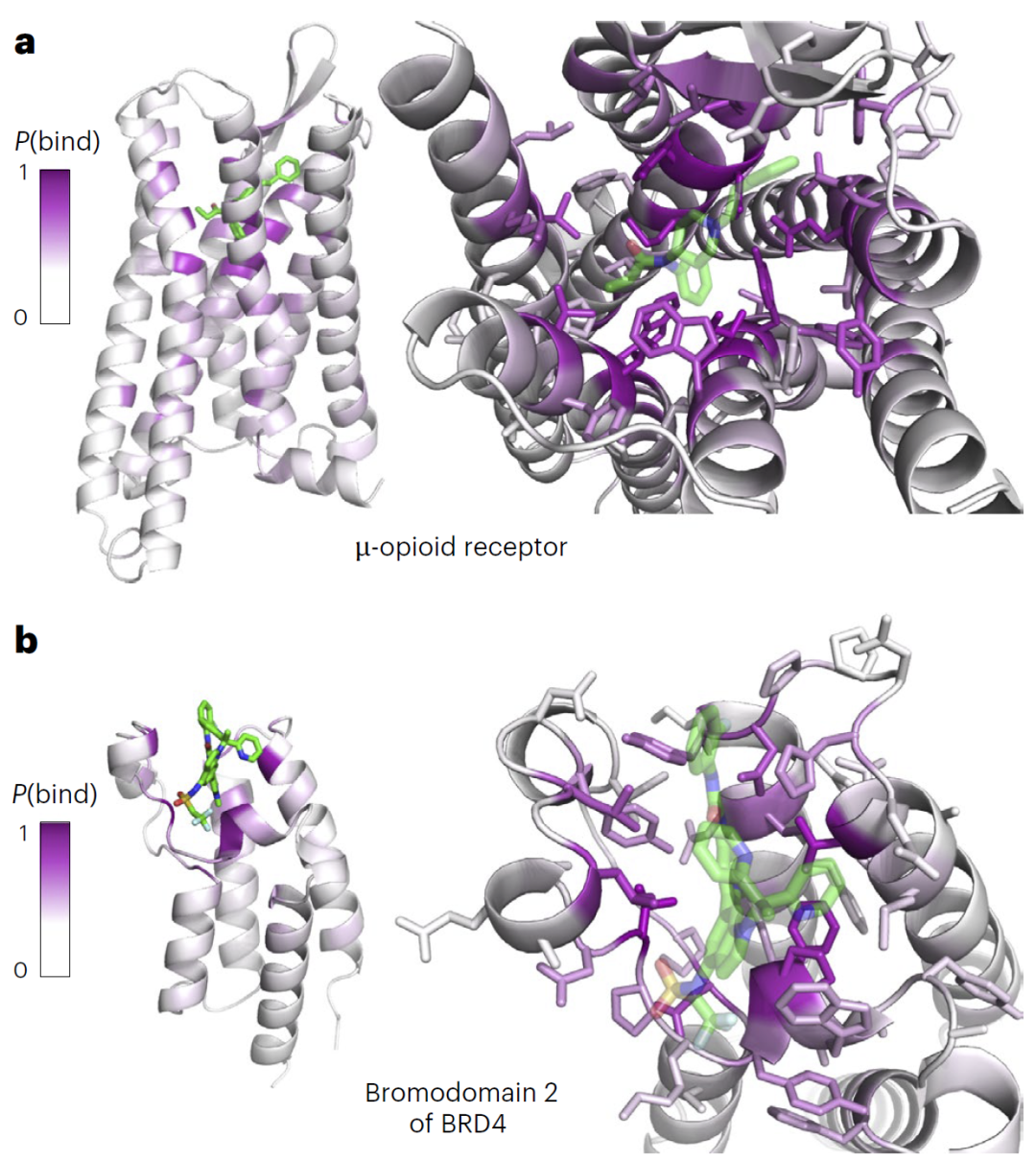
图4|AF2BIND在未参与训练的蛋白质上的预测表现。
诱饵氨基酸激活与配体化学性质的关系
由于AF2BIND模型能够追踪不同诱饵氨基酸对预测结果的贡献,研究人员进一步分析这些激活模式与真实配体性质之间的关系。
研究发现,疏水氨基酸诱饵的激活程度与配体的疏水性呈正相关,而极性氨基酸诱饵则更容易在亲水配体结合位点中被激活。这表明AF2BIND不仅可以识别结合位点,还能够反映潜在配体的化学性质特征。
这种激活模式实际上提供了一种“化学指纹”,可用于推测结合口袋更适合结合何种类型的小分子。
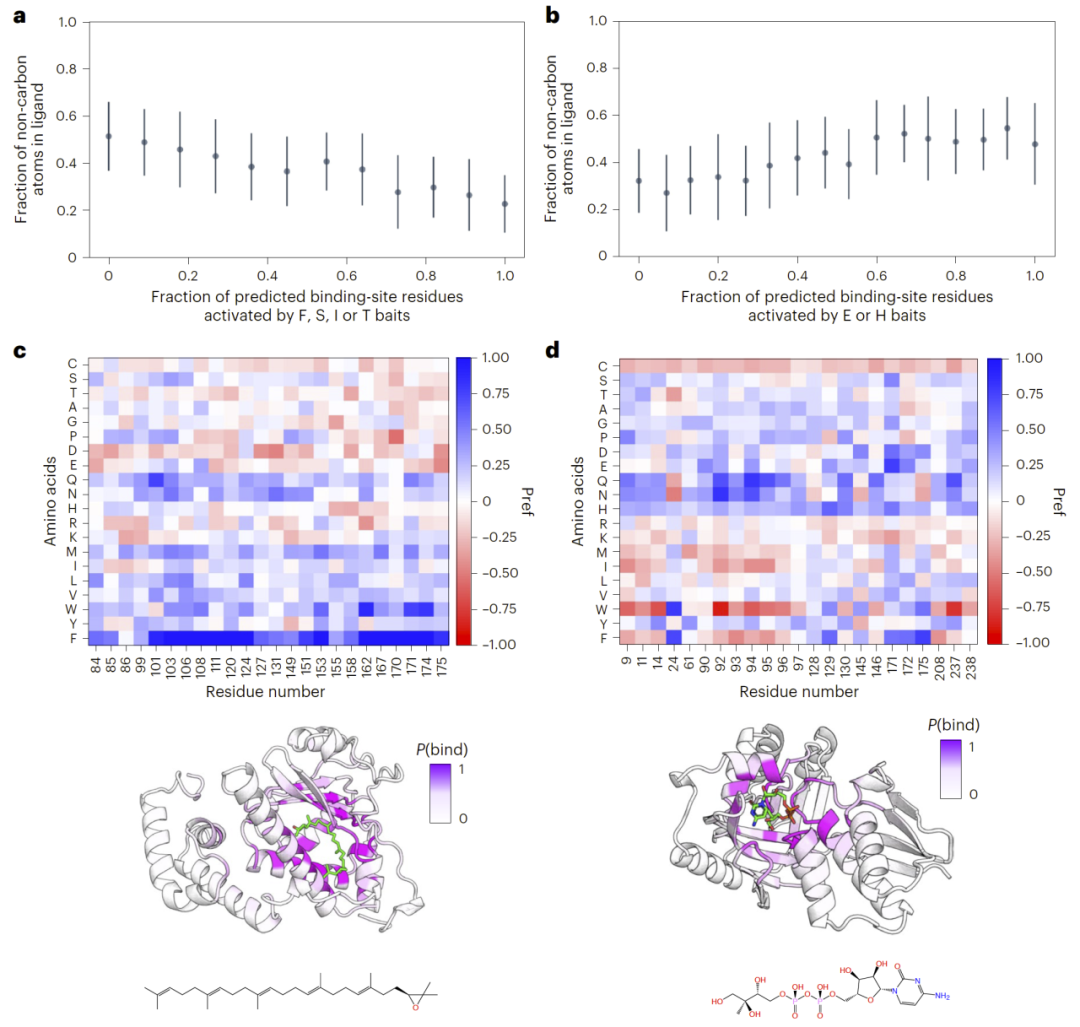
图5|诱饵氨基酸及其组合的激活模式与配体疏水性相关。
人类蛋白质组中的结合位点预测
研究人员将AF2BIND应用于AlphaFold数据库中的人类蛋白质结构,系统预测整个蛋白质组中的潜在小分子结合位点。
结果显示,在13,000多个蛋白中预测到超过20,000个潜在结合位点,其中大量位点并未被同源建模或传统口袋预测方法识别。与常用口袋检测工具P2Rank相比,两种方法在部分预测上互补,共同识别出近30,000个潜在结合位点。
许多AF2BIND预测位点在药物可及性评分上表现良好,表明这些位点可能具有药物开发潜力。
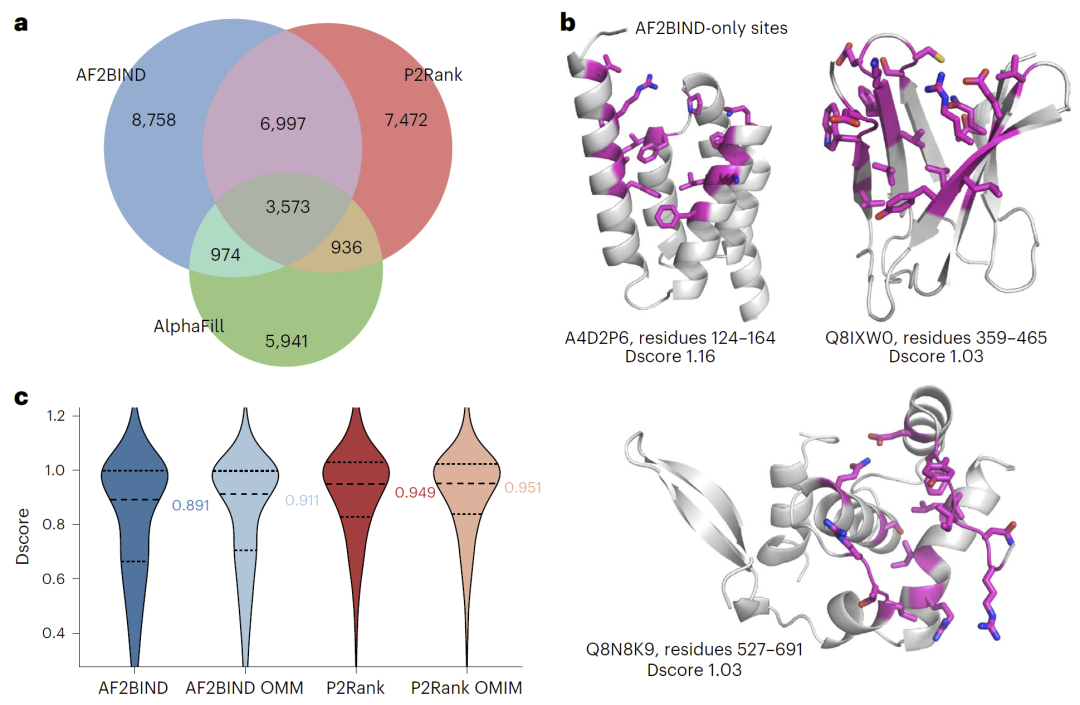
图6|AF2BIND在整个人类蛋白质组中预测到传统同源建模(AlphaFill)和P2Rank未识别的潜在可成药结合位点。
讨论
本研究表明,AlphaFold2在结构预测过程中学习到的内部表示包含丰富的功能信息,可用于识别蛋白质中的小分子结合位点。通过简单的逻辑回归模型,AF2BIND成功将这些表示转化为高效的结合位点预测工具。
与传统口袋检测方法相比,AF2BIND不仅能够识别典型口袋结构,还能够预测更浅表或更分散的结合位点,这些位点可能与蛋白–蛋白、蛋白–核酸或蛋白–肽相互作用相关,并可能成为潜在的小分子调控靶点。
该方法在整个人类蛋白质组上的应用表明,大量潜在药物结合位点仍有待探索。未来,将AF2BIND与配体对接、共结构预测以及化学蛋白质组学数据结合,有望进一步推动药物靶点发现和功能位点解析。
整理 | DrugOne团队
参考资料
Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods (2026).
https://doi.org/10.1038/s41592-026-03011-2

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号