生信星球day7-毽子
原创

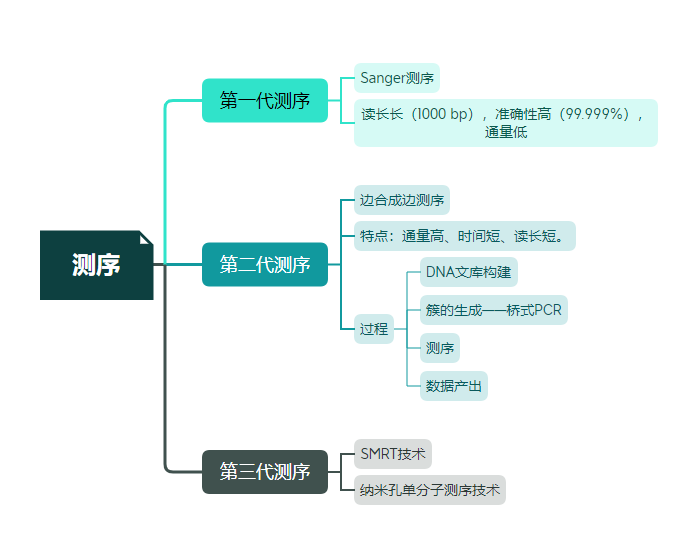
测序原理
我感觉这个讲得挺好的:
【中英双语】Illumina测序原理详解 | 边合成边测序
素材来源:YouTube官方 https://www.youtube.com/watch?v=fCd6B5HRaZ8 from一只小蛮要
Fastq格式:一种基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式,一般都包含有4行。
第一行:由‘@’开始,后面跟着序列ID和可选的描述,序列ID是唯一的;
第二行:碱基序列;
第三行:由‘+’开始,后面是序列的描述信息;
第四行:第二行序列的质量评价(quality value)。

from 生信星球

Fasta格式:

from 生信星球
1:以“>”为开头,fasta格式标志。
2:序列ID号,gi号,NCBI数据库的标识符,具有唯一性。
格式为:gi|gi号|来源标志|序列标志(接收号、名称等),若某项缺失可以留空,“|”保留。
3:序列描述。
4:碱基序列,序列中允许空格、换行、空行,一般一行60个。
Fastq文件→Fasta文件
Linux命令
法1:sed '/^@/!d;s//>/;N' your.fastq > your.fasta
法2:seqtk seq -A input.fastq > output.fasta
FASTX-Toolkit
•一款用于处理Short-Reads FASTA/FASTQ文件的程序,里面包含了丰富的Fasta/Fastq文件格式转换、统计等命令。
http://hannonlab.cshl.edu/fastx_toolkit/
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号