基于Kallisto或Salmon的转录组定量流程
原创
基于Kallisto或Salmon的转录组定量流程
原创
生信学习者
发布于 2024-06-13 09:18:52
发布于 2024-06-13 09:18:52
欢迎大家关注全网生信学习者系列:
- WX公zhong号:生信学习者
- Xiao hong书:生信学习者
- 知hu:生信学习者
- CDSN:生信学习者2
介绍
Kallisto和Salmon在RNA-seq数据分析中,相比于包含hisat2和STAR等软件的流程,展现出更高的处理速度。这主要归因于它们基于转录组序列reference(即cDNA序列)的特性和k mer比对原理。以下是关于Kallisto和Salmon在RNA-seq流程中速度优势的关键点归纳:
- 基于转录组序列reference:Kallisto和Salmon直接利用转录组序列(即cDNA序列)作为比对参考,而不是完整的基因组。这种策略显著减少了比对过程中需要考虑的序列范围,因此提高了处理速度。
- k mer比对原理:这两款软件都采用了k mer比对原理,即将RNA-seq数据中的读段(reads)切分成较短的k-mer片段,并与转录组参考序列进行比对。这种方法避免了传统比对方法中的复杂性和计算量,进一步提高了处理速度。
- 研究RNA-seq过程基因变化:当研究者希望了解RNA-seq过程中基因表达的变化,且不需要检测新的转录本时,Kallisto和Salmon提供了快速获取转录本丰度信息的解决方案。它们的快速和准确性使得它们成为RNA-seq数据分析中的常用工具。
Kallisto和Salmon基于转录组序列reference和k mer比对原理的设计,使得它们在RNA-seq数据分析中展现出显著的速度优势,特别是在不需要检测新转录本的情况下,能够快速地获取转录本的丰度信息。
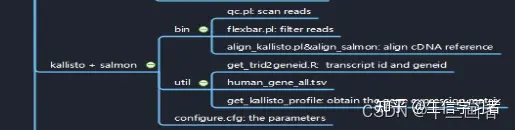
安装软件
conda install -c bioconda kallisto salmon -y过滤原始 fastq files
任何RNA-seq流程都需要对原始fastq reads文件进行质控,质控包括去除接头、barcode等额外引入的序列,当然也包括舍弃低质量的reads。这类软件有,cutadapt、flexbar和trimmomatic等。
download reference
转录本序列可以从多个网站下载,这里我们下载ENSEMBL数据库的小鼠cDNA数据。
wget ftp://ftp.ensembl.org/pub/release-100/fasta/mus_musculus/cdna/Mus_musculus.GRCm38.cdna.all.fabuild index
kallisto index -i kallisto_index/Mus_musculus.GRCm38 Mus_musculus.GRCm38.cdna.all.fa
salmon index -i salmon_index/Mus_musculus.GRCm38 -t Mus_musculus.GRCm38.cdna.all.fa -p 20生成批量运行的脚本
准备文件:samples.path.tsv(原始数据文件)
SampleID | LaneID | Path |
|---|---|---|
HF_NC01 | HF_NC01_RNA_b2.r2 | /01.rename/HF_NC01_RNA_b2.r2.fq.gz |
HF_NC01 | HF_NC01_RNA_b2.r1 | /01.rename/HF_NC01_RNA_b2.r1.fq.gz |
HF_NC02 | HF_NC02_RNA_b2.r1 | /01.rename/HF_NC02_RNA_b2.r1.fq.gz |
HF_NC02 | HF_NC02_RNA_b2.r2 | /01.rename/HF_NC02_RNA_b2.r2.fq.gz |
HF_NC03 | HF_NC03_RNA_novo.r2 | /01.rename/HF_NC03_RNA_novo.r2.fq.gz |
HF_NC03 | HF_NC03_RNA_novo.r1 | /01.rename/HF_NC03_RNA_novo.r1.fq.gz |
过滤文件目录:02.trim (高质量的reads文件)
撰写脚本quant_feature.py:生成批量运行shell文件
# key command
kallisto quant
-i kallisto_index/Mus_musculus.GRCm38
-g gtf_file
-o kallisto_quant/HF07
-b 30
-t 10 02.trim/HF07_1.fastq.gz 02.trim/HF07_2.fastq.gz
salmon quant
-i salmon_index/Mus_musculus.GRCm38/
-l IU -p 10
-1 02.trim/HF07_1.fastq.gz
-2 02.trim/HF07_2.fastq.gz
-o salmon_quant/HF07
--numBootstraps 30#!/usr/bin/python
import os
import re
import sys
import argparse as ap
from collections import defaultdict
current_abosulte_path = os.getcwd()
def parse_arguments(args):
parser = ap.ArgumentParser(description= "the initial script of pipeline")
parser.add_argument('-t','--trim',metavar='<string>', type=str, help='trimmed data dir')
parser.add_argument('-s','--sample',metavar='<string>', type=str, help='SampleID|LaneID|Path')
parser.add_argument('-o','--output',metavar='<string>', type=str, help='the output directory')
parser.add_argument('-g','--gtf',metavar='<string>', type=str, help='the genome annotation file')
parser.add_argument('-i','--index',metavar='<string>', type=str, help='the index of genome')
parser.add_argument('-p','--prefix',metavar='<string>', type=str, help='the prefix of output file')
return parser.parse_args()
def create_folder(folder):
'''
create folder for the result
'''
folder_path = os.path.join(current_abosulte_path, folder)
if not os.path.exists(folder_path):
os.makedirs(folder_path)
return folder_path
def get_path_file(sample):
'''
the connections among sampleid, laneid and path
'''
dict_lane = defaultdict(list)
with open(sample, 'r') as f:
for line in f:
line = line.strip()
if not line.startswith("SampleID"):
group = re.split(r'\s+', line)
dict_lane[group[0]].append(group[1])
return dict_lane
def generate_shell(trim_folder, sample_file, outdir, gtf_file, genome_index, file_name):
trim_file_folder = os.path.join(current_abosulte_path, trim_folder)
output_path = create_folder(outdir)
output_name = os.path.join(current_abosulte_path, file_name)
sample_name = get_path_file(sample_file)
output_f = open(output_name, 'w')
for key in sample_name:
out_sample_prefix = os.path.join(output_path, key)
#target1 = [x for x in sample_name[key] if re.findall(r'r1', x)]
#fq1 = os.path.join(trim_file_folder, ("_".join([target1[0], "val_1.fq.gz"])))
#target2 = [x for x in sample_name[key] if re.findall(r'r2', x)]
#fq2 = os.path.join(trim_file_folder, ("_".join([target2[0], "val_2.fq.gz"])))
fq1 = os.path.join(trim_file_folder, ("_".join([key, "1.fastq.gz"])))
fq2 = os.path.join(trim_file_folder, ("_".join([key, "2.fastq.gz"])))
if os.path.isfile(fq1) and os.path.isfile(fq2):
fq = " ".join([fq1, fq2])
if re.findall(r'kallisto', genome_index):
shell = " ".join(["kallisto quant -i", genome_index, "-g", \
"gtf_file", "-o", out_sample_prefix, "-b 30 -t 10", fq])
elif re.findall(r'salmon', genome_index):
shell = " ".join(["salmon quant -i", genome_index, \
"-l IU -p 10", \
"-1", fq1, "-2", fq2, \
"-o", out_sample_prefix,\
"--numBootstraps 30"])
output_f.write(shell + "\n")
output_f.close()
def main():
args = parse_arguments(sys.argv)
generate_shell(args.trim, args.sample, args.output, args.gtf, args.index, args.prefix)
if __name__ == '__main__':
main()运行脚本: 生成RUN文件
# kallisto
python quant_feature.py -t 02.trim/ -s 46RNA.samples.path.tsv -o kallisto_quant -g /disk/share/database/Mus_musculus.GRCm38_release100/Mus_musculus.GRCm38.cdna.all.fa -i /disk/share/database/Mus_musculus.GRCm38_release100/kallisto_index/Mus_musculus.GRCm38 -p RUN.kallisto_quant.sh
# salmon
python quant_feature.py -t 02.trim/ -s 46RNA.samples.path.tsv -o salmon_quant -g /disk/share/database/Mus_musculus.GRCm38_release100/Mus_musculus.GRCm38.cdna.all.fa -i /disk/share/database/Mus_musculus.GRCm38_release100/salmon_index/Mus_musculus.GRCm38/ -p RUN.salmon_quant.sh获取expression matrix:
撰写get_merged_table.R,从quant目录获取整合所有样本表达值的matrix文件
#!/bin/usr/R
library(dplyr)
library(argparser)
library(tximport)
# parameter input
parser <- arg_parser("merge transcript abundance files") %>%
add_argument("-s", "--sample",
help = "the sampleID files") %>%
add_argument("-d", "--dir",
help = "the dir of output") %>%
add_argument("-g", "--gene",
help = "transcriptID into geneID") %>%
add_argument("-t", "--type",
help = "the type of software") %>%
add_argument("-c", "--count",
help = "the method of scale for featurecounts") %>%
add_argument("-n", "--name",
help = "the name of transcript file") %>%
add_argument("-o", "--out",
help = "the merged files's dir and name")
args <- parse_args(parser)
# prepare for function
sample <- args$s
dir <- args$d
tx2gen <- args$g
type <- args$t
scount <- args$c
name <- args$n
prefix <- args$o
tx2gene_table <- read.csv(tx2gen, header=T)
tx2gene <- tx2gene_table %>%
dplyr::select(tx_id, gene_id, gene_name)
phen <- read.table(sample, header=T, sep="\t")
sample_name <- unique(as.character(phen$SampleID))
files <- file.path(dir, sample_name, name)
names(files) <- sample_name
result <- tximport(files,
type = type,
countsFromAbundance = scount,
tx2gene = tx2gene,
ignoreTxVersion = T)
save(result, file = prefix)Rscript get_merged_table.R \
-s samples.path.tsv \
-d kallisto_quant/ \
-g EnsDb.Mmusculus.v79_transcriptID2geneID.csv \
-t kallisto \
-c no \
-n abundance.tsv \
-o kallisto_quant.RData
Rscript get_merged_table.R \
-s samples.path.tsv \
-d salmon_quant/ \
-g EnsDb.Mmusculus.v79_transcriptID2geneID.csv \
-t salmon \
-c no \
-n quant.sf \
-o salmon_quant.RData获取TPM和FPKM表达矩阵:可在本地电脑运行
library(dplyr)
library(tibble)
load("kallisto_quant.RData")
phen <- read.csv("phenotype.csv")
kallisto_count <- round(result$counts) %>% data.frame()
kallisto_length <- apply(result$length, 1, mean) %>% data.frame() %>%
setNames("Length")
get_profile <- function(matrix, exon_length,
y=phen,
occurrence=0.2,
ncount=10){
# matrix=count_format
# exon_length=count_length
# occurrence=0.2
# ncount=10
# filter with occurrence and ncount
prf <- matrix %>% rownames_to_column("Type") %>%
filter(apply(dplyr::select(., -one_of("Type")), 1,
function(x){sum(x > 0)/length(x)}) > occurrence) %>%
data.frame(.) %>%
column_to_rownames("Type")
prf <- prf[rowSums(prf) > ncount, ]
# change sampleID
sid <- intersect(colnames(prf), y$SampleID)
phen.cln <- y %>% filter(SampleID%in%sid) %>%
arrange(SampleID)
prf.cln <- prf %>% dplyr::select(as.character(phen.cln$SampleID))
# determine the right order between profile and phenotype
for(i in 1:ncol(prf.cln)){
if (!(colnames(prf.cln)[i] == phen.cln$SampleID[i])) {
stop(paste0(i, " Wrong"))
}
}
colnames(prf.cln) <- phen.cln$SampleID_v2
# normalizate the profile using FPKM and TPM
exon_length.cln <- exon_length[as.character(rownames(prf.cln)), , F]
for(i in 1:nrow(exon_length.cln)){
if (!(rownames(exon_length.cln)[i] == rownames(prf.cln)[i])) {
stop(paste0(i, " Wrong"))
}
}
dat <- prf.cln/exon_length.cln$Length
dat_FPKM <- t(t(dat)/colSums(prf.cln)) * 10^9
dat_TPM <- t(t(dat)/colSums(dat)) * 10^6
dat_count <- prf.cln[order(rownames(prf.cln)), ]
dat_fpkm <- dat_FPKM[order(rownames(dat_FPKM)), ]
dat_tpm <- dat_TPM[order(rownames(dat_TPM)), ]
res <- list(count=dat_count, fpkm=dat_fpkm, tpm=dat_tpm)
return(res)
}
prf_out <- function(result, type="STAR"){
dir <- "../../Result/Profile/"
if(!dir.exists(dir)){
dir.create(dir)
}
count_name <- paste0(dir, type, "_filtered_counts.tsv")
FPKM_name <- paste0(dir, type, "_filtered_FPKM.tsv")
TPM_name <- paste0(dir, type, "_filtered_TPM.tsv")
write.table(x = result$count,
file = count_name,
sep = '\t',
quote = F,
col.names = NA)
write.table(x = result$fpkm,
file = FPKM_name,
sep = '\t',
quote = F,
col.names = NA)
write.table(x = result$tpm,
file = TPM_name,
sep = '\t',
quote = F,
col.names = NA)
}
kallisto_prf <- get_profile(kallisto_count, kallisto_length)
# output
prf_out(kallisto_prf, type = "kallisto")Reference
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号