
简化DNA/三元加密过程
简化DNA/三元加密过程
提问于 2022-02-04 18:13:24
我想简化DNA/三元加密算法。一方面,我有一个加密过程,我把一组碱基-3数三叉(0,1,2)转化为DNA核苷酸(A,T,C,G)。而去加密的过程正好相反。
这种加密的要点是没有两个相同的连续核苷酸在一起。因此,即使最初的载体是c(0,0,0,0),加密的DNA也不会是c("A","A","A","A")。这是通过遵循表的算法来实现的。
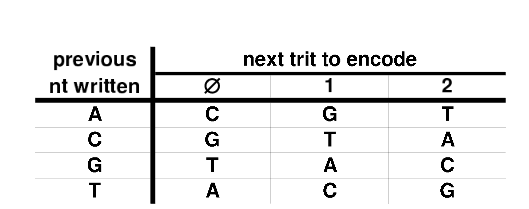
在哪里处理第一个三位一体/核苷酸,你假设前病毒核苷酸是"A"。以免看到几个例子。
加密: c(0,0,0,0) "A")
- De-encrypting c("C","G","T",“A”)
- De-encrypting c("T","A","C","G") -> c(2,0,0,0)
)
这就是我如何实现流程自动化的方法:
加密
> S4
[1] "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "2" "0" "2" "1" "0" "2" "1" "2"
[31] "1" "1" "0" "1" "0" "1" "1" "0" "0" "0" "1" "0" "1" "2" "1" "0" "0" "0" "0" "0"
S5 <-c(1:length(S4))
for (i in 1:length(S4)) {
if (i == 1 ) {
if (S4[1] == "0") {
S5[1] <- "C"
}
if (S4[1] == "1") {
S5[1] <- "G"
}
if (S4[1] == "2") {
S5[1] <- "T"
}
}
if (i != 1) {
if (S5[i-1] == "A" && S4[i] == "0") {
S5[i] <- "C"
}
if (S5[i-1] == "A" && S4[i] == "1") {
S5[i] <- "G"
}
if (S5[i-1] == "A" && S4[i] == "2") {
S5[i] <- "T"
}
if (S5[i-1] == "C" && S4[i] == "0") {
S5[i] <- "G"
}
if (S5[i-1] == "C" && S4[i] == "1") {
S5[i] <- "T"
}
if (S5[i-1] == "C" && S4[i] == "2") {
S5[i] <- "A"
}
if (S5[i-1] == "G" && S4[i] == "0") {
S5[i] <- "T"
}
if (S5[i-1] == "G" && S4[i] == "1") {
S5[i] <- "A"
}
if (S5[i-1] == "G" && S4[i] == "2") {
S5[i] <- "C"
}
if (S5[i-1] == "T" && S4[i] == "0") {
S5[i] <- "A"
}
if (S5[i-1] == "T" && S4[i] == "1") {
S5[i] <- "C"
}
if (S5[i-1] == "T" && S4[i] == "2") {
S5[i] <- "G"
}
}
}
> S5
[1] "CGTACGTACGTACGTACGTACGCGCTATCAGACTAGACGTCGATCGTACG"De-encryption
adn <- S5
adn <- unlist(strsplit(adn, split = ""))
adn
s4 <- c()
for (i in 1:length(adn)) { #Loops to transform DNA sequence to terminary according to manual
if (i != 1) {
if (adn[i-1] == "A" && adn[i] == "C") {
s4[i] <- 0
}
if (adn[i-1] == "A" && adn[i] == "G") {
s4[i] <- 1
}
if (adn[i-1] == "A" && adn[i] == "T") {
s4[i] <- 2
}
if (adn[i-1] == "C" && adn[i] == "G") {
s4[i] <- 0
}
if (adn[i-1] == "C" && adn[i] == "T") {
s4[i] <- 1
}
if (adn[i-1] == "C" && adn[i] == "A") {
s4[i] <- 2
}
if (adn[i-1] == "G" && adn[i] == "T") {
s4[i] <- 0
}
if (adn[i-1] == "G" && adn[i] == "A") {
s4[i] <- 1
}
if (adn[i-1] == "G" && adn[i] == "C") {
s4[i] <- 2
}
if (adn[i-1] == "T" && adn[i] == "A") {
s4[i] <- 0
}
if (adn[i-1] == "T" && adn[i] == "C") {
s4[i] <- 1
}
if (adn[i-1] == "T" && adn[i] == "G") {
s4[i] <- 2
}
}
if (i == 1 ) {
if (adn[1] == "C") {
s4[1] <- 0
}
if (adn[1] == "G") {
s4[1] <- 1
}
if (adn[1] == "T") {
s4[1] <- 2
}
}
}
> s4
[1] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 2 1 0 2 1 2 1 1 0 1 0 1 1 0 0 0 1 0 1 2 1 0 0 0 0 0问题
我该如何使这几步更优雅呢?
回答 1
Stack Overflow用户
回答已采纳
发布于 2022-02-04 18:47:52
我们可以通过制作一个包含编码的data.frame来简化这一点,这样我们就可以在不对每个组合进行测试的情况下访问它们:
enc <- data.frame(A=c('C','G','T'),
C=c('G','T','A'),
G=c('T','T','C'),
T=c('A','C','G'),
row.names = c(0,1,2))解码功能很简单,因为我们有我们需要的所有预先信息。我们只需要遍历字符串,获取当前基和前一个基(如果没有,则默认为'A' ),然后使用它们查找表中与这些值匹配的位置,返回具有trit值的行名:
decode <- function(d) {
sapply(seq_along(d), function(i) {
base = d[i]
prev = ifelse(length(d[i-1]) == 0, 'A', d[i-1])
rownames(enc)[which(enc[[prev]] == base)]
})
}
decode(c('T', 'A', 'C', 'G'))
[1] 2 0 0 0编码有点困难,因为我们需要查看以前编码的值,但它几乎是一样的。唯一的区别是,我们需要使用for循环并在编码时将每个值存储在列表中,这样我们就可以回顾并找到前面的trit。
encode <- function(e) {
res <- list()
for (i in seq_along(e)) {
trit = e[i]
prev = ifelse(length(res[i-1]) == 0, 'A', res[[i-1]])
res[[i]] <- enc[as.character(trit), prev] # Select row by name, not index
}
return(unlist(res)) # unlist to return vector, not list
}
encode(c(0,0,0,0))
[1] "C" "G" "T" "A"页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/70991010
复制相关文章
相似问题
腾讯云开发者
