
分子动力学模拟:波动偶极子模型的实现
我在对二氧化硅进行分子动力学模拟。一段时间前,我转向了波动的偶极子模型,经过了很大的努力,我仍然难以实现它。
简而言之,系统中的所有氧原子都是可极化的,它们的偶极矩取决于它们相对于系统中所有其它原子的位置。更特别的是,我使用TS势(http://digitallibrary.sissa.it/bitstream/handle/1963/2874/tangney.pdf?sequence=2),其中偶极子在每一时间步骤都被迭代地发现。
这意味着,在计算作用在原子上的力时,我必须考虑到这个势能依赖于坐标。以前,我使用简单的成对势模型,所以我会设置程序,用通过微分势能表达式得到的解析公式来计算力。
现在我不知所措:如何实现这一新的潜力?在我发现的所有文章中,它们只给出了公式,但没有给出算法。在我看来,当我计算一个原子上的力时,我必须考虑这个原子的偶极子的变化,所有相邻原子的偶极子的变化,然后再考虑更多原子的偶极子的变化,等等,因为它们相互依赖。毕竟,正是由于这种相互依赖关系,偶极子才会在每一时间步骤中被迭代地发现。显然,我不能迭代计算每个原子的力,因为算法的计算复杂度太高了。我应该用一些简单的函数来解释偶极子的变化吗?这看起来也不是一个好主意,因为偶极子是迭代计算的,精度很高,然后,当它真正重要的地方(计算力),我们会使用粗糙的函数?
那么我如何实现这个模型呢?另外,是否有可能像我以前那样解析地计算力,或者是否有必要使用导数的有限差分公式来计算它们?
我还没有在文献中找到我的问题的答案,但如果你知道一些文章,网站,或书籍,这些材料的突出显示,请指导我到那个来源。
谢谢您抽时间见我!
==================================================================================
更新:
谢谢你的回答。不幸的是,这不是我的问题。我没有问如何计算偶极子,而是如何计算力,因为这些偶极子随着运动而有很大的变化。
我试图以简单的方式计算力(没有考虑到偶极子之间的距离相互依赖,只需计算每个步骤上的偶极子,然后像那些偶极子是静态的那样计算这些力),但是我得到的结果在物理上是不正确的。
为了分析这种情况,我建立了一个由两个原子组成的系统的模拟: Si和O,它们有相反的电荷,所以它们振荡。能量时间依赖图如下所示:
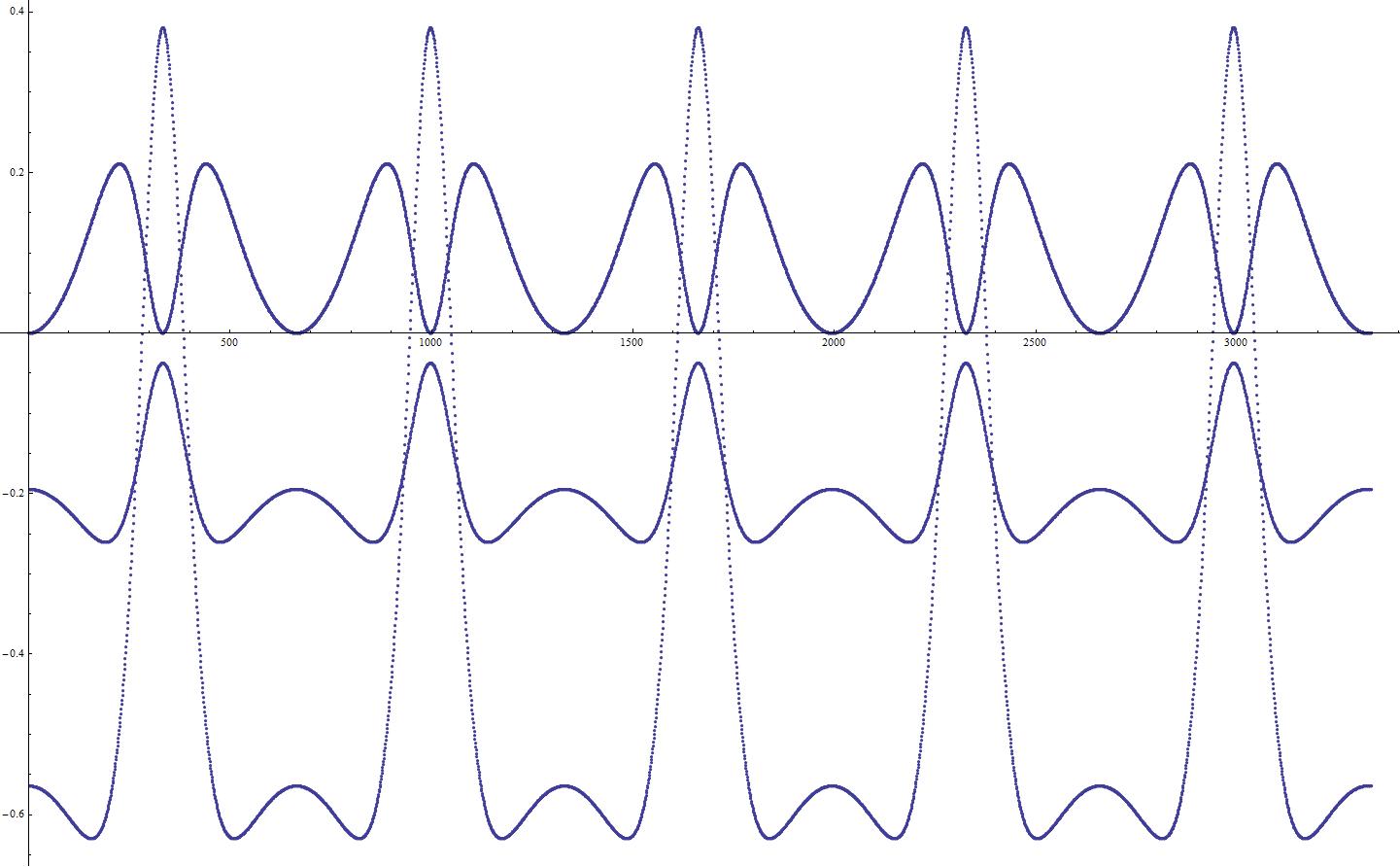
顶部的曲线代表动能,中间的曲线表示势能而不考虑偶极子相互作用,底部的曲线代表系统的势能,其中考虑了偶极子相互作用。
从这张图可以清楚地看到,系统正在做它不应该做的事情:爬上潜在的斜坡。所以我决定这是因为我没有考虑偶极矩,坐标依赖。例如,在给定的时间点,我们计算力,它们被定向,使两个原子相互移动。但是当我们把它们移向对方时,偶极矩就会发生变化,我们会发现我们最终得到的势能比以前更高了!在下一阶段,情况是一样的。
所以问题是,如何考虑这一影响,因为我所能想到的任何一种方法,要么计算过于激烈,要么过于粗糙。
回答 1
Stack Overflow用户
发布于 2013-05-23 15:53:44
我不确定我是否完全理解您的问题,但听起来您可能需要实现马尔可夫链类型的解决方案?
有关更多信息,请参见这篇漂亮的文章:http://freakonometrics.hypotheses.org/6803
编辑。我认为这是因为听起来你有一个系统,每个原子的状态取决于它的邻居,而邻居的状态又取决于它们的邻居等等。从概念上讲,这可以建模为一个巨大的矩阵,并且您可以基于它的邻居(?)迭代地更新每个值。这是一个棘手的问题,但本文介绍了如何使用马尔可夫链来解决一个很大的转移矩阵问题,而不是计算实际的矩阵。
https://stackoverflow.com/questions/16686252
复制相似问题
腾讯云开发者
