转录组测序分析专题——比对/定量
原创

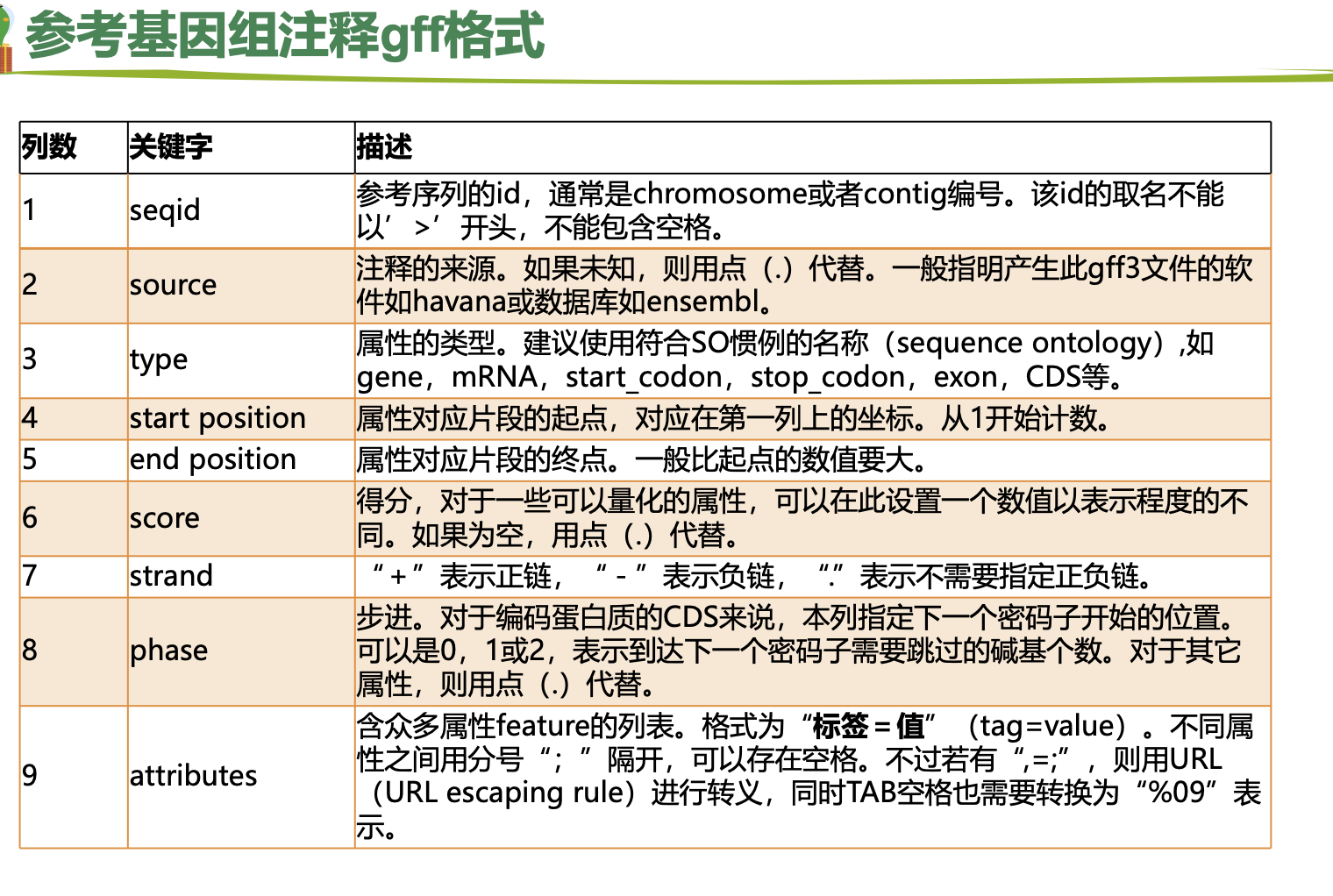
一、参考基因组准备
基因组文件:fasta
注释文件:gff/gtf
1.常用的参考基因组数据库
Ensembl:www.ensembl.org
NCBI:https://www.ncbi.nlm.nih.gov/projects/genome/gu ide/human/index.shtml
UCSC:http://www.genome.ucsc.edu/
2.参考基因组下载

参考基因组
## 参考基因组准备:注意参考基因组版本信息
# 下载,Ensembl:http://asia.ensembl.org/index.html
# http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/
# 进入到参考基因组目录
mkdir -p $HOME/database/GRCh38.105
cd $HOME/database/GRCh38.105
# 下载基因组序列axel curl
nohup wget -c http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz >dna.log &
# 下载转录组序列
nohup wget -c http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz >rna.log &
# 下载基因组注释文件
nohup wget -c http://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.chr.gtf.gz >gtf.log &
nohup wget -c http://ftp.ensembl.org/pub/release-105/gff3/homo_sapiens/Homo_sapiens.GRCh38.105.chr.gff3.gz >gff.log&
# 上述文件下载完整后,再解压;否则文件不完整就解压会报错
# 再次强调,一定要在文件下载完后再进行解压!!!
nohup gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz Homo_sapiens.GRCh38.cdna.all.fa.gz >unzip.log &二、fasta数据格式介绍


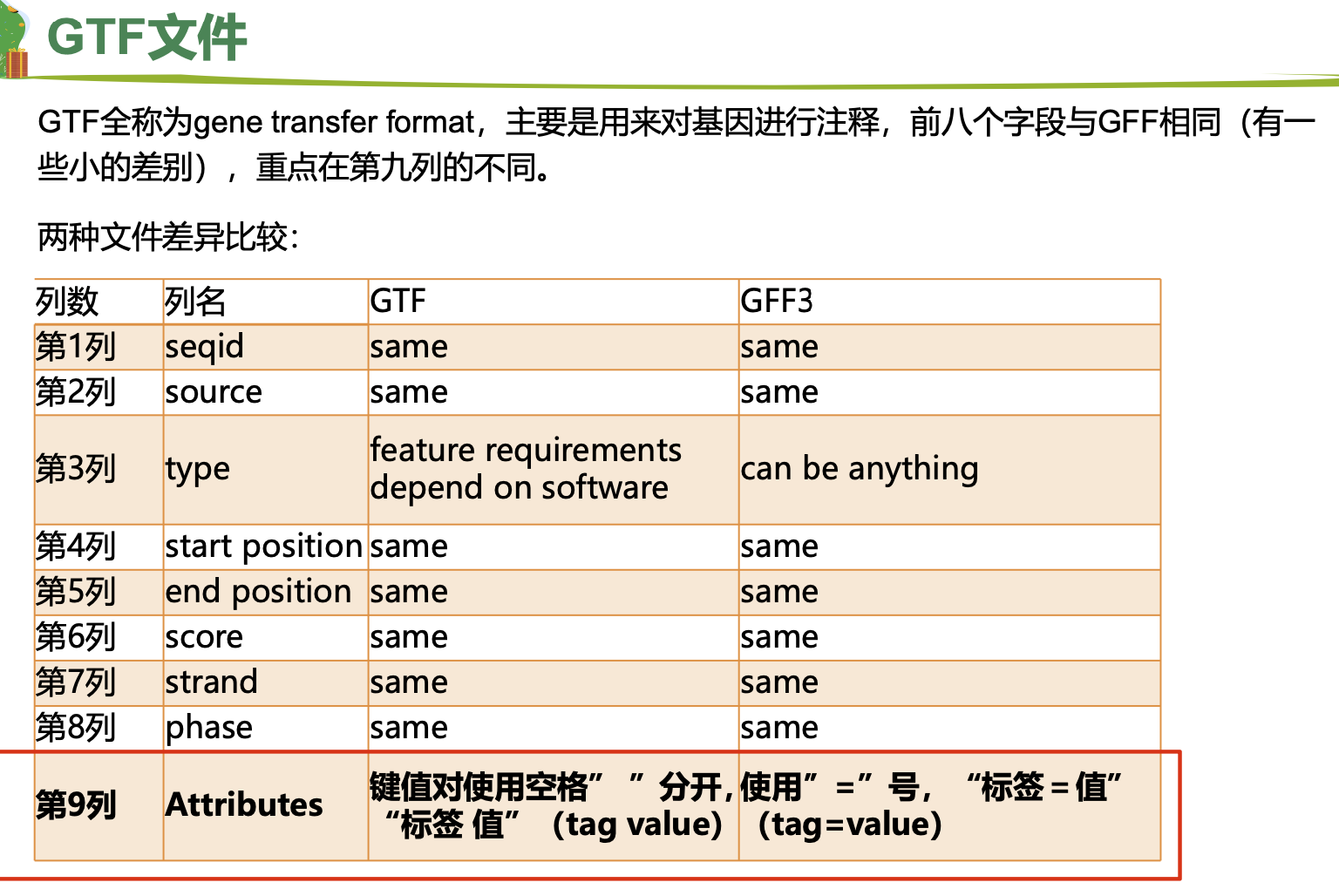
三、gtf/gff文件介绍

gtf和gff的第三列和第九列内容不一样

习题:
1.fastq与fasta文件转换
# 答案1
zless -S SRR1039511_1_val_1.fq.gz |awk '{ if(NR%4==1){print">" substr($0,2)} if(NR%4==2){print} }' | less -S
# 答案2
zless -S SRR1039510_1_val_1.fq.gz |paste - - - - |cut -f 1,2 |tr '@' '>' |tr '\t' '\n' |less -S2.从gff或者gft文件中获取基因的ID与symbol对应关系,以及biotype类型
应用:ID与symbol转换本地化,不依赖于第三方工具和软件包,并可以根据biotype类型区分mRNA,lncRNA以及miRNA等信息。
# 从gff或者gft文件中获取ID与symbol对应关系,以及biotype类型
zless -S Homo_sapiens.GRCh38.104.chr.gtf.gz |awk -F'\t' '{if($3=="gene"){print$9}}' |awk -F';' '{print$1,$3,$5}' |awk '{print$2"\t"$4"\t"$6}' |sed 's/"//g' |grep 'protein_coding' >protein_coding_id2name.xls四、数据比对
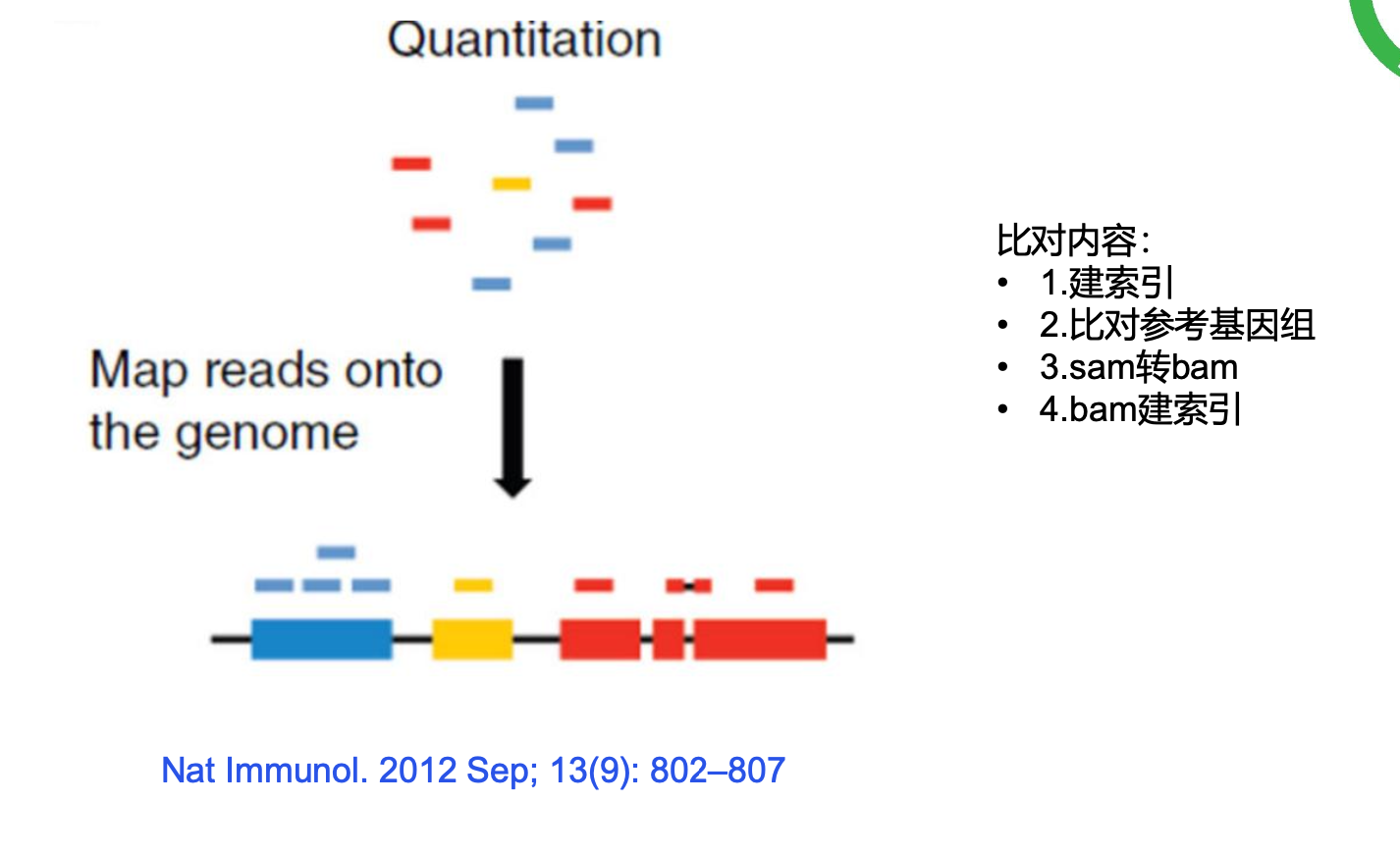


1.hisat2比对
## ----构建索引
# 进入参考基因组目录
cd $HOME/database/GRCh38.105
# Hisat2构建索引,构建索引时间比较长,建议提交后台运行,一般会运行20多分钟左右
## 后续索引可直接使用服务器上已经构建好的进行练习
# vim Hisat2Index.sh
mkdir Hisat2Index
fa=Homo_sapiens.GRCh38.dna.primary_assembly.fa
fa_baseName=GRCh38.dna
hisat2-build -p 12 -f ${fa} Hisat2Index/${fa_baseName}
# 运行
nohup sh Hisat2Index.sh >Hisat2Index.sh.log &
## ----比对
# 进入比对文件夹
cd $HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
## 单个样本比对,步骤分解
index=/home/t_rna/database/GRCh38.104/Hisat2Index/GRCh38.dna
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore/
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
hisat2 -p 10 -x ${index} \
-1 ${inputdir}/SRR1039510_1_val_1.fq.gz \
-2 ${inputdir}/SRR1039510_2_val_2.fq.gz \
-S ${outdir}/SRR1039510.Hisat_aln.sam
# sam转bam
samtools sort -@ 15 -o SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sam
# 对bam建索引
samtools index SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sorted.bam.bai
# 多个样本批量进行比对,排序,建索引
# Hisat.sh内容: 注意命令中的-,表示占位符,表示|管道符前面的输出。
## 此处索引直接使用服务器上已经构建好的进行练习
# vim Hisat.sh
index=/home/t_rna/database/GRCh38.104/Hisat2Index/GRCh38.dna
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore/
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
cat ../../data/cleandata/trim_galore/ID | while read id
do
hisat2 -p 5 -x ${index} -1 ${inputdir}/${id}_1_val_1.fq.gz -2 ${inputdir}/${id}_2_val_2.fq.gz 2>${id}.log | samtools sort -@ 3 -o ${outdir}/${id}.Hisat_aln.sorted.bam - && samtools index ${outdir}/${id}.Hisat_aln.sorted.bam ${outdir}/${id}.Hisat_aln.sorted.bam.bai
done
# 统计比对情况
multiqc -o ./ SRR*log
# 提交后台运行
nohup sh Hisat.sh >Hisat.log &1.比对结果文件:sam/bam格式

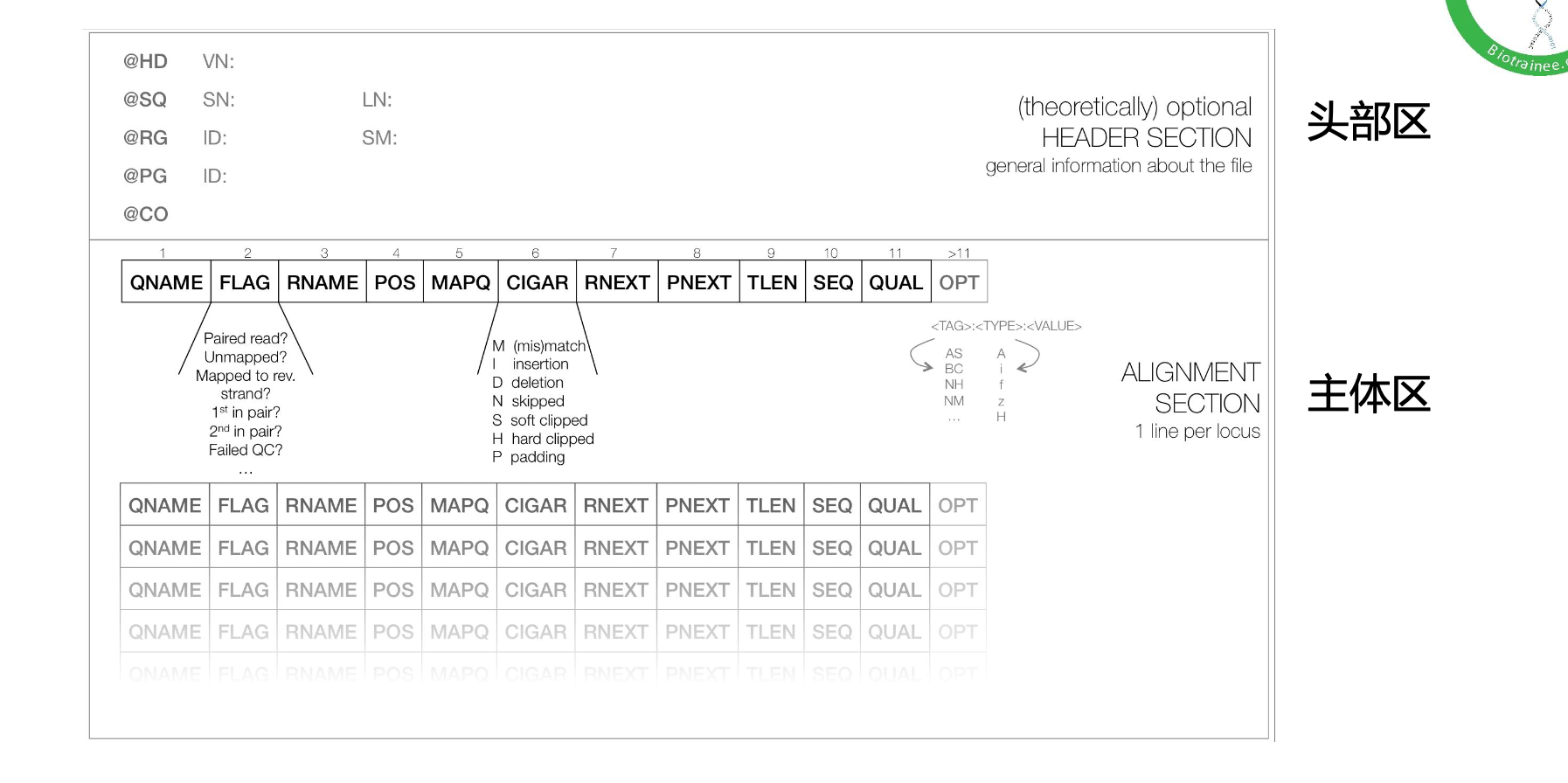

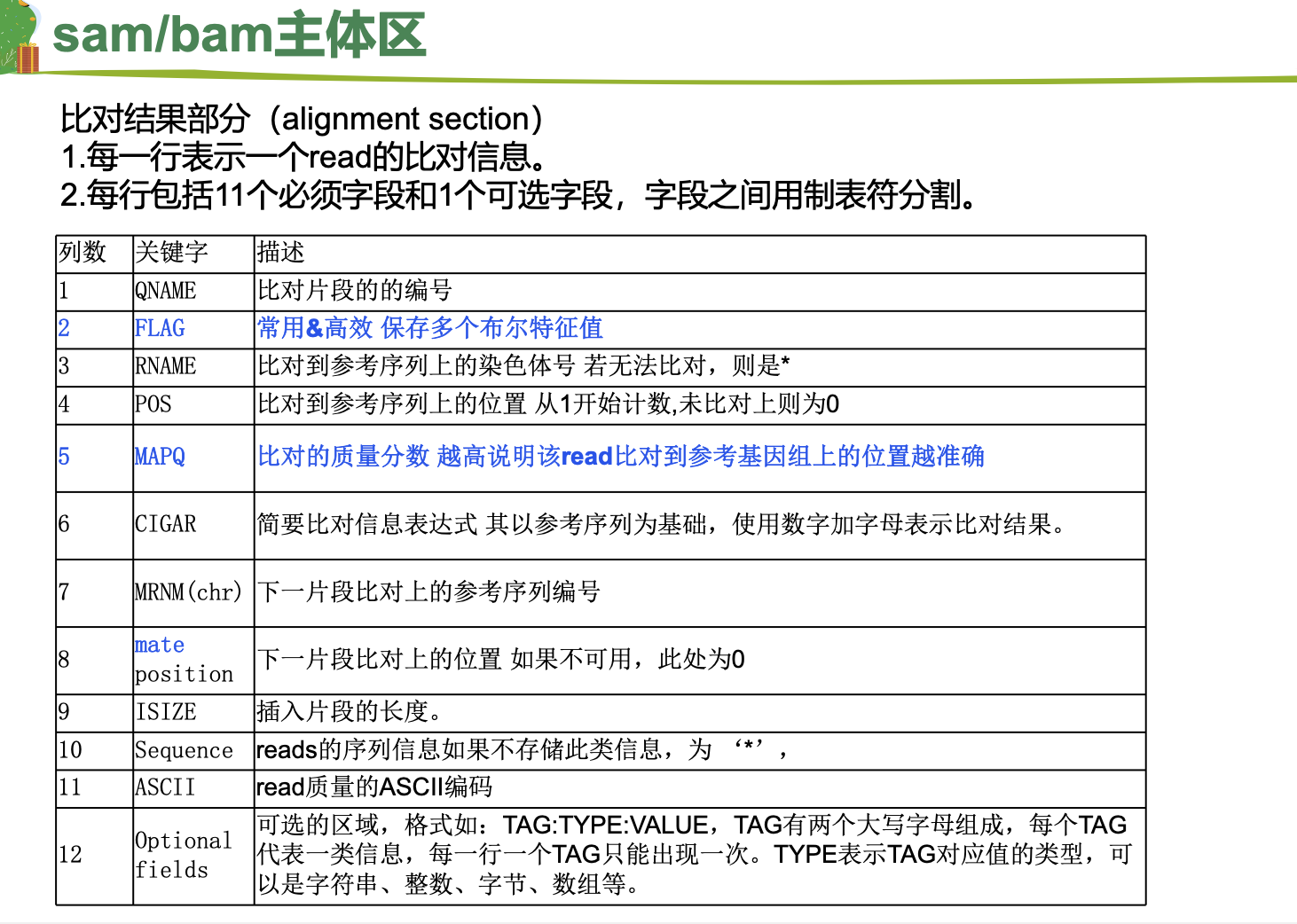


2.sam/bam文件查看

#samtools查看bam文件
samtools view [file.bam] | less -S #查看bam
samtools view -H file.bam |less -S #查看头部区
samtools view -h file.bam |less -S #查看全部2.subjunc比对——除了RNA-seq比对还能做外显子junction分析
## ----构建索引
# 进入参考基因组目录
cd $HOME/database/GRCh38.105
# subjunc构建索引,构建索引时间比较长大约40分钟左右,建议携程sh脚本提交后台运行
## 后续索引可直接使用服务器上已经构建好的进行练习
# vim SubjuncIndex.sh
mkdir SubjuncIndex
fa=Homo_sapiens.GRCh38.dna.primary_assembly.fa
fa_baseName=GRCh38.dna
subread-buildindex -o SubjuncIndex/${fa_baseName} ${fa}
# 运行
nohup sh SubjuncIndex.sh >SubjuncIndex.sh.log &
## ----比对
# 进入文件夹
cd $HOME/project/Human-16-Asthma-Trans/Mapping/Subjunc
# vim subjunc.sh
index=/home/t_rna/database/GRCh38.104/SubjuncIndex/GRCh38.dna
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Subjunc
cat ../../data/cleandata/trim_galore/ID | while read id
do
subjunc -T 10 -i ${index} -r ${inputdir}/${id}_1_val_1.fq.gz -R ${inputdir}/${id}_2_val_2.fq.gz -o ${outdir}/${id}.Subjunc.bam 1>${outdir}/${id}.Subjunc.log 2>&1 && samtools sort -@ 6 -o ${outdir}/${id}.Subjunc.sorted.bam ${outdir}/${id}.Subjunc.bam && samtools index ${outdir}/${id}.Subjunc.sorted.bam ${outdir}/${id}.Subjunc.sorted.bam.bai
done
# 运行
nohup sh subjunc.sh >subjunc.log &3.sam/bam应用
统计比对结果
# 单个样本
samtools flagstat -@ 3 SRR1039510.Hisat_aln.sorted.bam
# 多个样本,vim flagstat.sh
ls *.sorted.bam | while read id
do
samtools flagstat -@ 10 ${id} > ${id/bam/flagstat}
done
# 质控
multiqc -o ./ *.flagstat
# 运行
nohup sh flagstat.sh >flagstat.log &samtools工具使用
##----view查看bam文件
samtools view SRR1039510.Hisat_aln.sorted.bam
samtools view -H SRR1039510.Hisat_aln.sorted.bam
samtools view -h SRR1039510.Hisat_aln.sorted.bam
##----index对bam文件建索引
samtools index SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sorted.bam.bai
##----flagstat统计比对结果
samtools flagstat -@ 3 SRR1039510.Hisat_aln.sorted.bam
##----sort排序 sam转bam并排序
samtools sort -@ 3 -o SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sam
##----depth统计测序深度
# 得到的结果中,一共有3列以指标分隔符分隔的数据,第一列为染色体名称,第二列为位点,第三列为覆盖深度
samtools depth SRR1039510.Hisat_aln.sorted.bam >SRR1039510.Hisat_aln.sorted.bam.depth.txt
##----计算某一个基因的测序深度
# 找到基因的坐标
zless -S Homo_sapiens.GRCh38.95.gff3.gz |awk '{if($3=="gene")print}' |grep 'ID=gene:ENSG00000186092' |awk '{print $1"\t"$4"\t"$5}' >ENSG00000186092.bed
samtools depth -b ENSG00000186092.bed SRR1039510.Hisat_aln.sorted.bam >ENSG00000186092.bed.depth
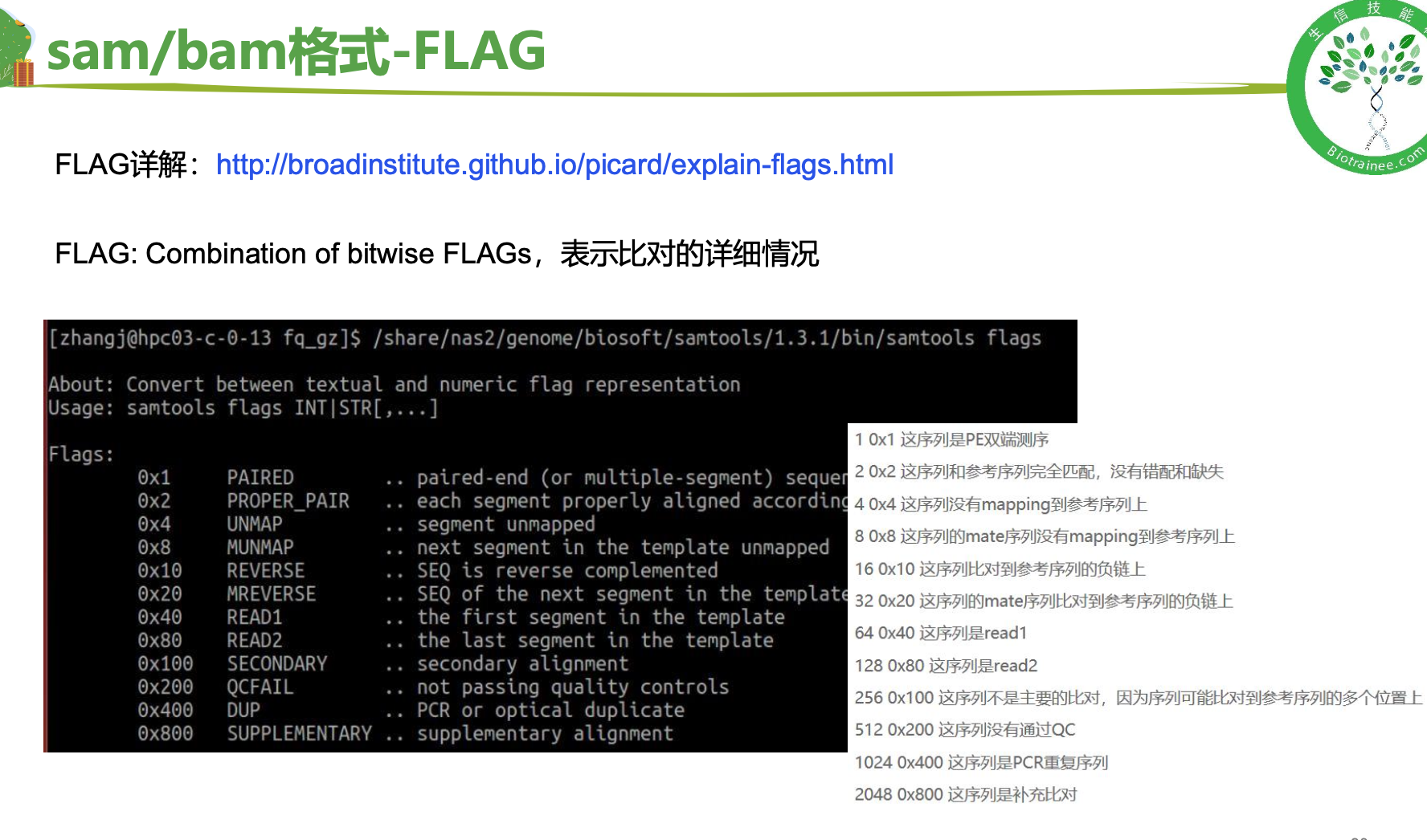
# 如何找到多比对的reads,flag值的理解
# (0x100) 代表着多比对情况,所以直接用samtools view -f 0x100可以提取 multiple比对的 情况五、表达定量
1.featureCounts



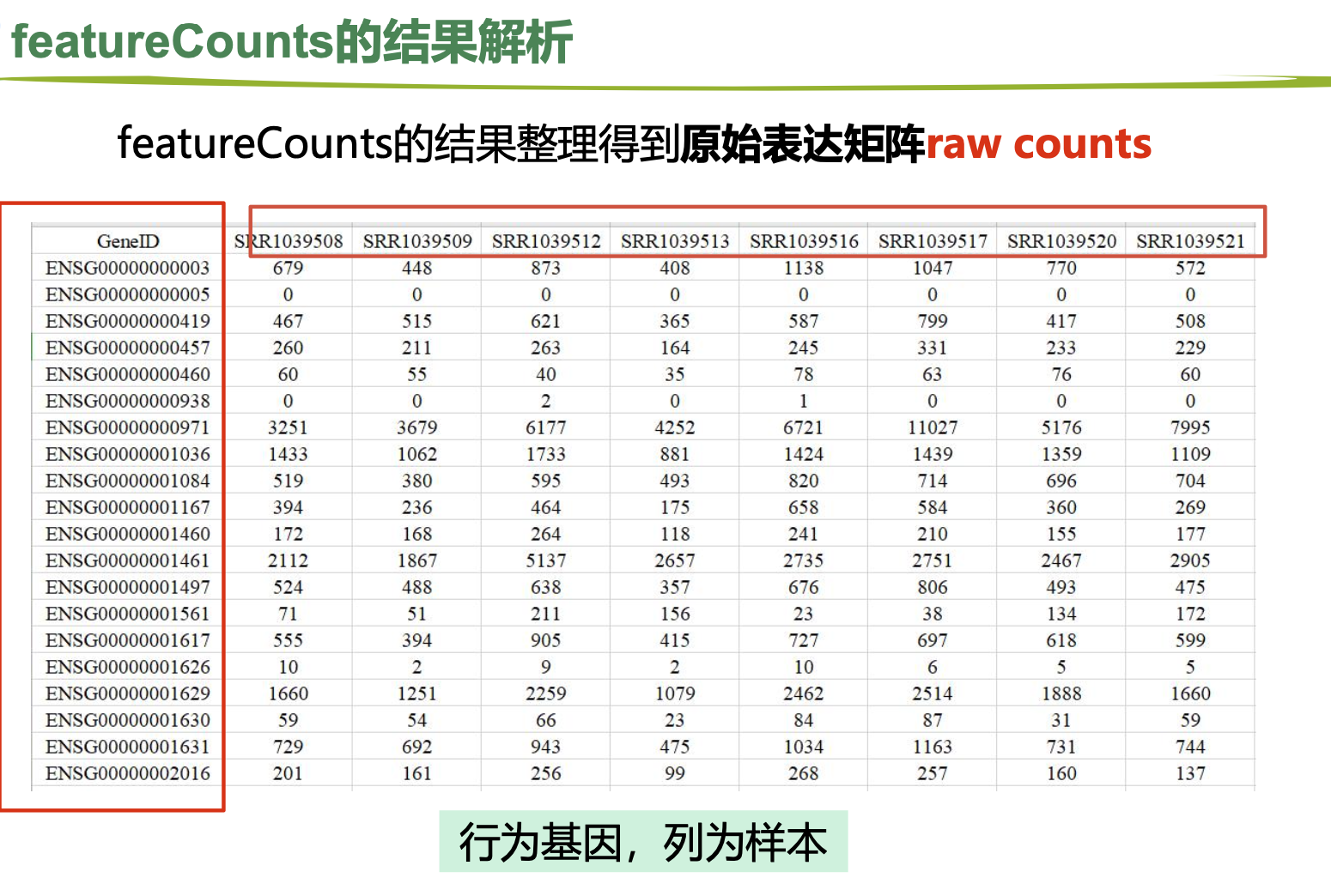
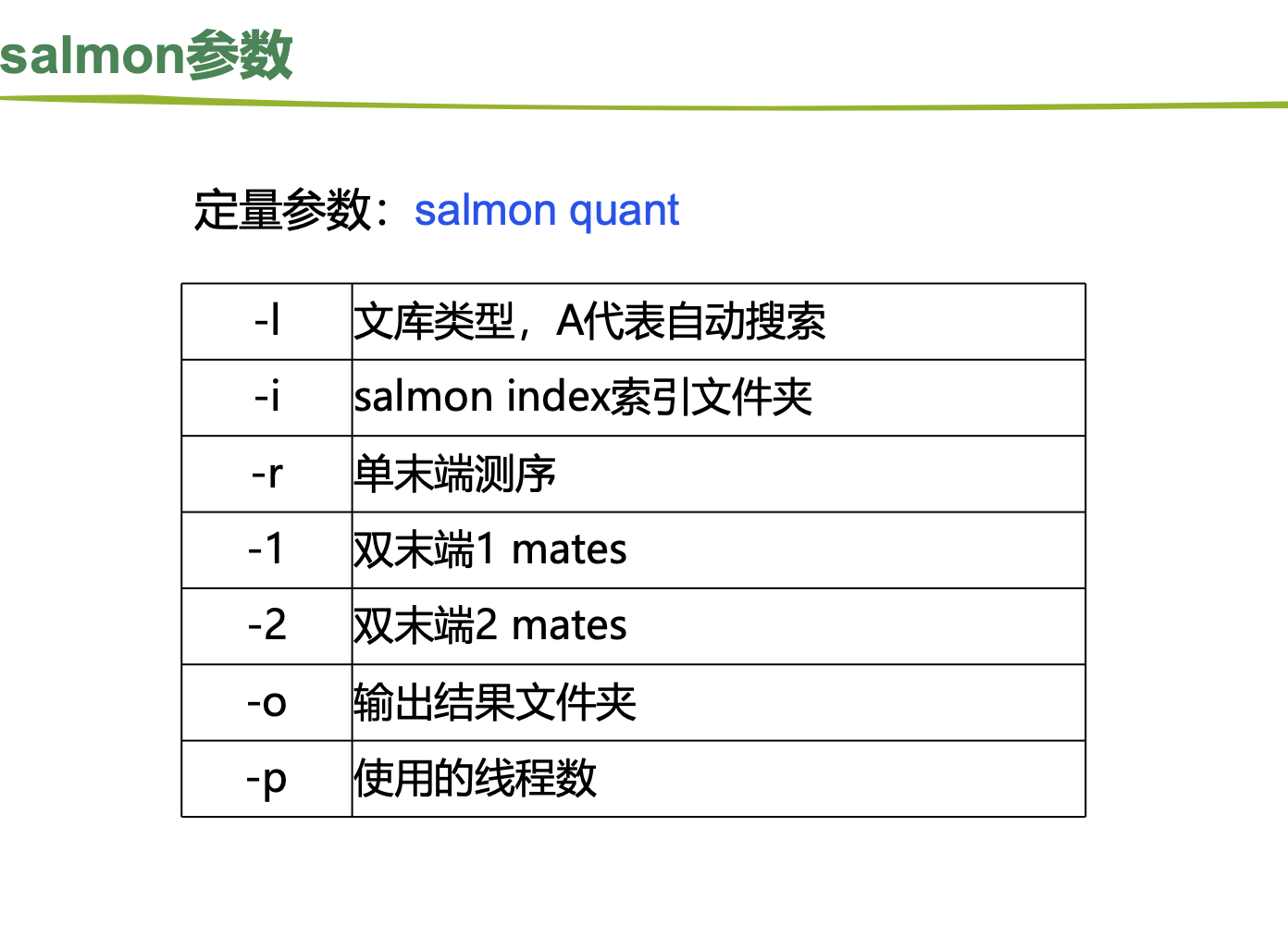
2.salmon
##----构建索引
## 后续索引可直接使用服务器上已经构建好的进行练习
cd $HOME/database/GRCh38.105
nohup salmon index -t Homo_sapiens.GRCh38.cdna.all.fa -i salmon_index >salmon-index.log &
cd $HOME/project/Human-16-Asthma-Trans/Expression/Salmon
##----运行
# 编写脚本,使用salmon批量对目录下所有fastq文件进行定量
# vim salmon.sh
index=/home/t_rna/database/GRCh38.104/salmon_index
input=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Expression/Salmon
cat ../../data/cleandata/trim_galore/ID |while read id
do
salmon quant -i ${index} -l A -1 ${input}/${id}_1_val_1.fq.gz -2 ${input}/${id}_2_val_2.fq.gz -p 5 -o ${outdir}/${id}.quant
done
##----合并表达矩阵
# 原始count值矩阵
# --quants:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{" $0 "}" }'
# --quants:ls -d *quant |sed ':a;N;s/\n/,/;t a;'|awk '{print "{"$0"}"}'
# --quants:ls -d *quant |xargs |tr ' ' ',' |awk '{print "{"$0"}"}'
# --names:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{"$0"}"}' |sed 's/.quant//g'
## 完整版:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{"$0"}"}' | perl -ne 'chomp;$a=$_;$a=~s/\.quant//g;print"salmon quantmerge --quants $_ --names $a --column numreads --output raw_count.txt \n";'
salmon quantmerge --quants {SRR1039510.quant,SRR1039511.quant,SRR1039512.quant} --names {SRR1039510,SRR1039511,SRR1039512} --column numreads --output raw_count.txt
# tpm矩阵
salmon quantmerge --quants {SRR1039510.quant,SRR1039511.quant,SRR1039512.quant} --names {SRR1039510,SRR1039511,SRR1039512} --column tpm --output tpm.txt
# 后台运行脚本
nohup sh salmon.sh >salmon.log &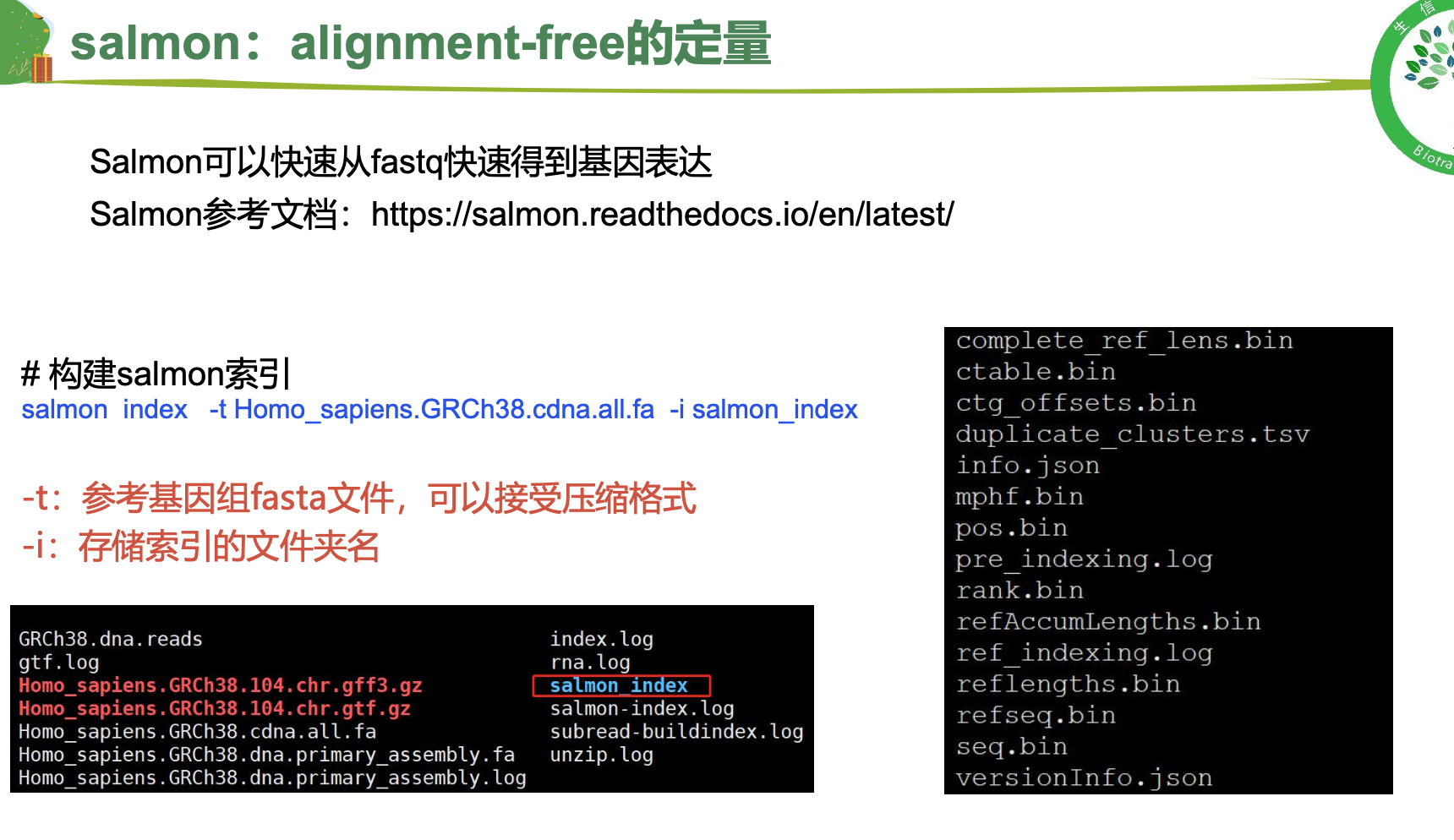

原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号