微生物全长16S | Full-length 16S Analysis -- PacBio Hifi Reads
原创
微生物全长16S | Full-length 16S Analysis -- PacBio Hifi Reads
原创
三代测序说
修改于 2024-02-09 11:41:18
修改于 2024-02-09 11:41:18
微生物研究新世代 -- 三代全长16S (Full-length 16S)
时至今日,微生物群落研究已全面进入测序分析阶段,当前研究主流处于二代扩增子与三代扩增子交接的时段。基于三代测序的菌群多样性组成谱分析能极大地提升物种分类鉴定的精确性和全面性,能更准确地还原样本中微生物群落的构成,实现“高分辨率”检测的同时,也为今后深入阐释菌群的代谢功能奠定了基础。
16S核糖体RNA(16S ribosomal RNA),简称16S rRNA,是原核生物核糖体中30S亚基的组成部分。16S rRNA基因存在于所有细菌的基因组中,长度约为1542 bp,包括 10 个保守区(Conserved region)和 9 个可变区(Variable region),保守区反映了物种间的亲缘关系,而可变区则反映了物种间的差异 (图1)。 16S rRNA基因,其分子大小适中,突变率小,是细菌系统分类研究中最有用的和最常用的分子标志。通过16S扩增子高通量测序,检测16S rDNA可变区的序列变异和丰度,可了解样品中微生物群落多样性和丰度信息,在微生物分类鉴定、微生态研究等方面起着重要的作用。
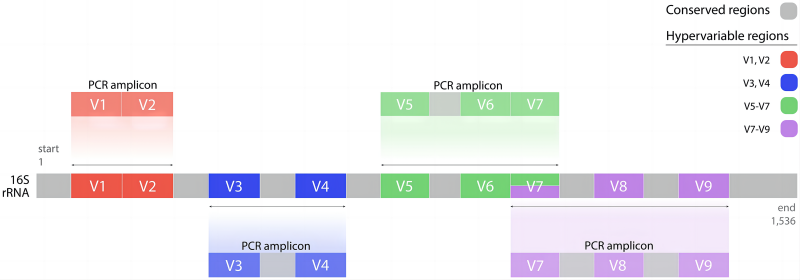
图1. 16S rRNA的结构和扩增区域
1990年,科学家们首次发现了环境样本中存在的16S rRNA序列(1),阐述了其研究潜力,自此开启了一个波澜壮阔的微生物群落研究时代。二代16S测序因其扩增片段极限长度仅500-600bp(双端overlap),因此对于二代扩增子挑选可变区是个大难题,挑选意味着妥协与信息丢失,如文章所示(3),属、种水平的未鉴定物种比例高 (图2)。三代16S扩增子测序,采用27F、1492R引物扩增全长片段(覆盖V1-V9区),则能够轻松覆盖16S总长约1500bp共9个可变区,最大程度保留了物种鉴定的可能性(图3)。
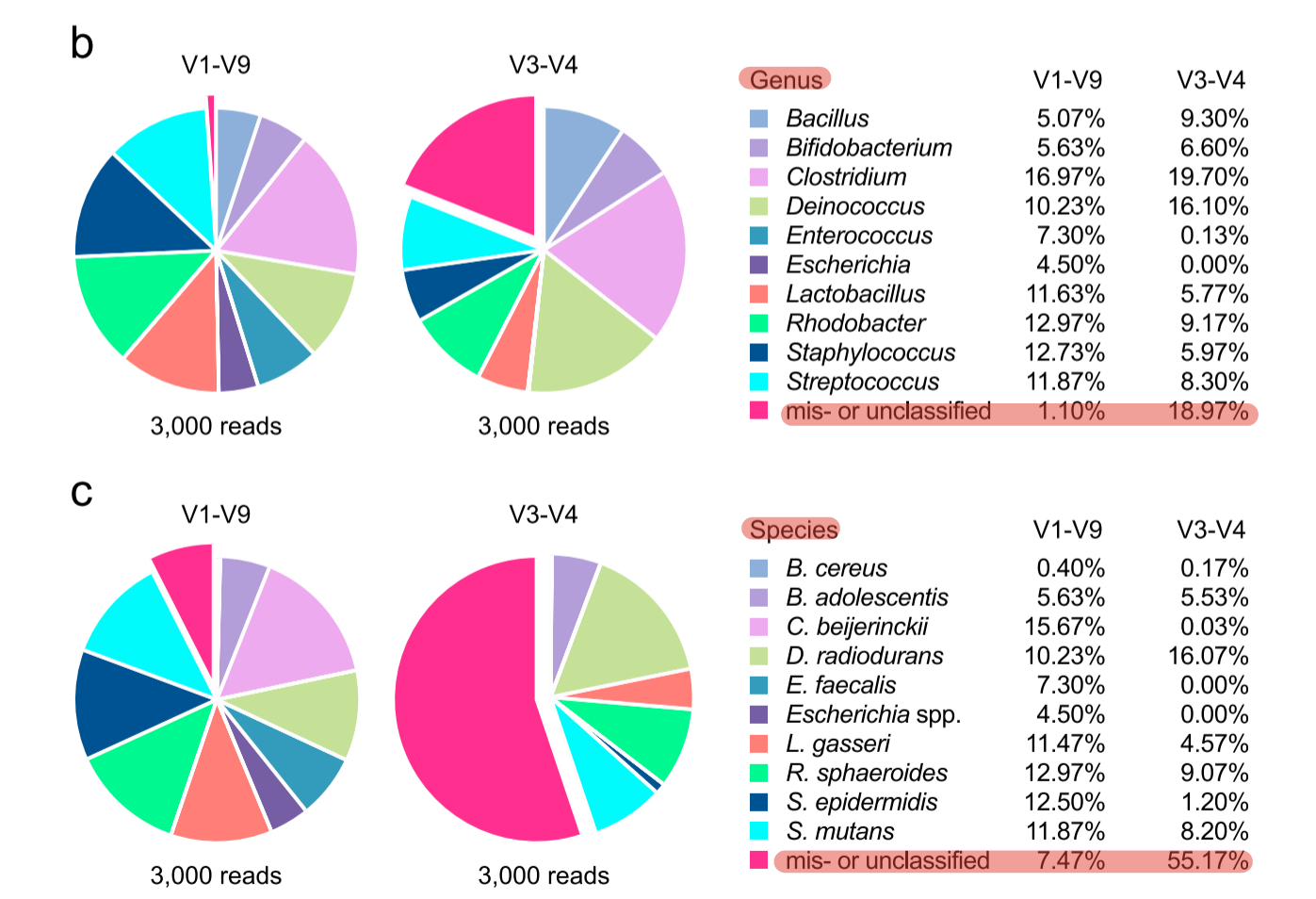
图2. 二代16s与三代全长16s未鉴定物种占比结果对比
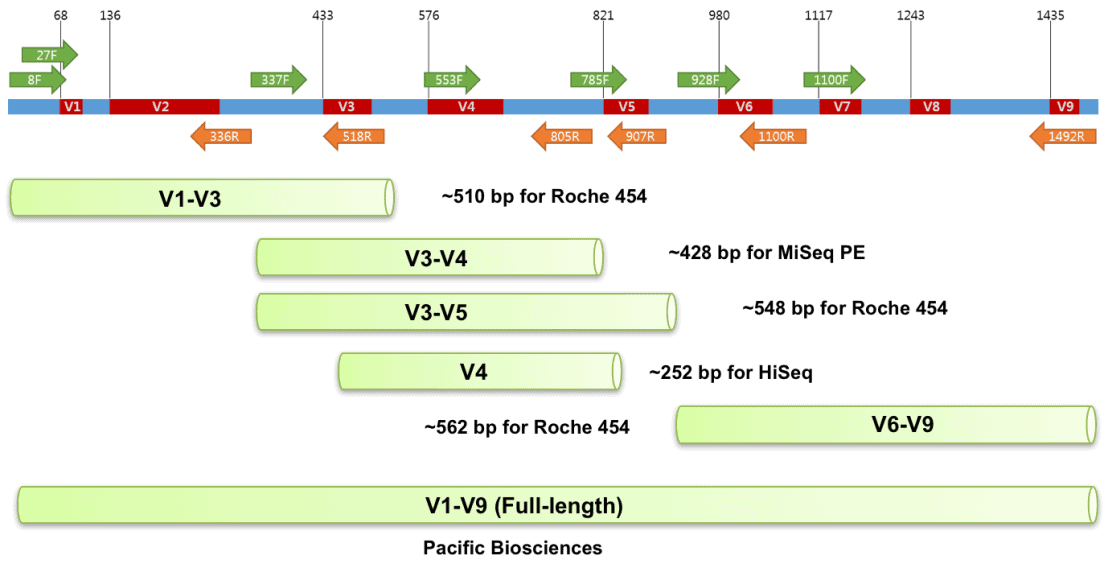
图3. 三代全长16S扩增区域
每一轮的技术革新都带来了研究思路上的改变,二代扩增子技术带来研究思路关注群落整体多样性变化、侧重门/属等水平微生物构成。三代扩增子技术则更进一步,更加关注不同组学间的关联,不仅关注门/属等水平物种丰度,也能够探索属内物种的协作/竞争关系 。有了如此高的分辨率表现,菌种级别的研究自然成为了研究重点,不同于过往对于二代16S科、属水平的研究,三代全长16S能够提供更全面且细致的菌株级别分析结果,让整个研究结果更贴近生态学功能,对于多组学关联以及后续课题实验指导、验证都有着巨大意义。从多组学关联的角度来看同样如此,更精细层面数据进行多组学关联往往能够揭示出更清晰的局部规律,这其中就有很多过往被忽略或无法触及的细节。
一、PacBio全长16S rRNA基因测序
PacBio全长16S rRNA基因测序采用27F、1492R引物扩增全长片段(覆盖V1-V9区),采用PacBio SMRT测序平台CCS(Circular Consensus Sequencing)模式进行测序分析。PacBio SMRT测序具有许多明显优势:
- 长读长,二代测序读长只能达到几百bp,而PacBio测序读长可达几十甚至上百kb。对于长度约为 1542bp 的16S rRNA基因,二代测序只能对部分区域如V4、V3V4、V4V5区进行测序,而PacBio测序可轻松跨越16S rRNA基因的全长序列。
- 高准确度,PacBio CCS模式获得的 HiFi Reads(High fidelity reads)自我矫正准确性高达99%以上, 兼顾测序数据长读长和高准确度。当测序酶读长达到 8Kb时,即可满足一条1.5Kb的16S rRNA基因序列循环矫正5次 (图4),最终获得高质量的16S全长序列。
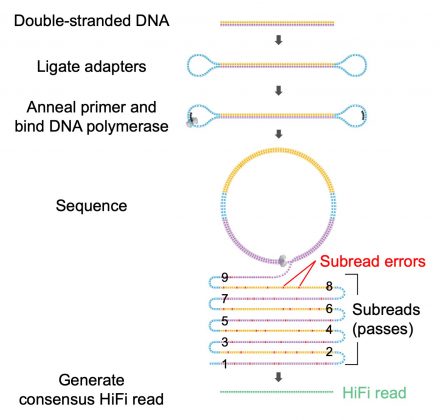
图4.CCS测序模式
- 测序过程无偏好,PacBio单分子实时(SMRT)测序不需要扩增步骤,可避免测序过程中引入的偏好,可较大程度上还原样本真实群落结构。
二、PacBio | HiFi Full-length 16S analysis 分析流程
HiFi Full-length 16S nextflow 分析流程旨在通过DADA2 和QIIME2将全长16S Hifi序列聚类为高质量的Amplicon Sequence Variants (ASVs),进而完成后续的分析 。此流程基于QIIME2,因此其能做的分析,如alpha多样性及beta多样性,物种注释和可视化,HiFi Full-length 16S分析流程均能够实现 (图5)。除了ASVs聚类,分析流程还能用vsearch进行OTU聚类。
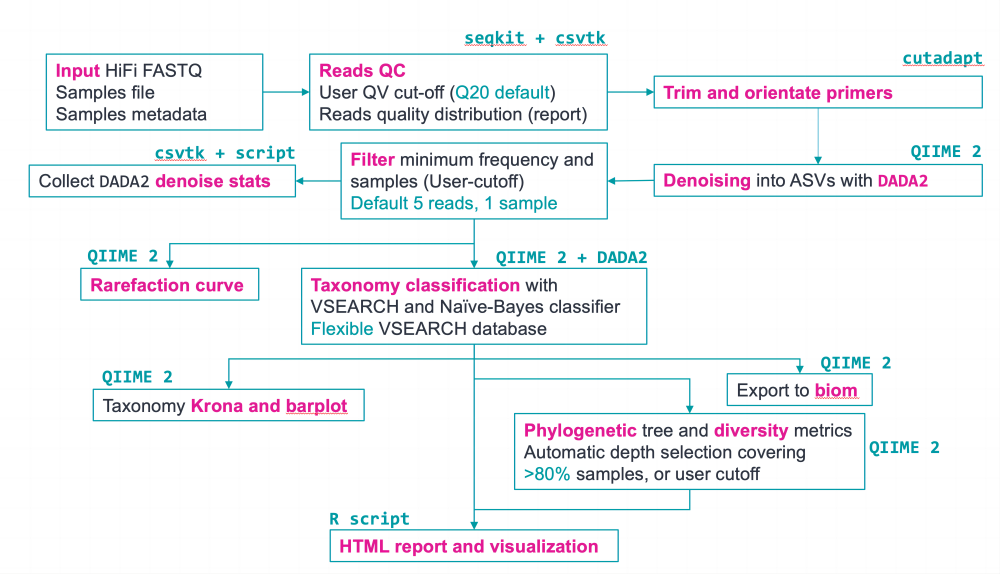
图5. HiFi Full-length 16S分析流程和产出结果文件
HiFi Full-length 16S 流程: https://github.com/PacificBiosciences/HiFi-16S-workflow
三、软件安装及测试
1. 从github上下载pb-16S-nt文件夹:
$ git clone https://github.com/PacificBiosciences/pb-16S-nf.git- 下载完成后,在当前路径会产生名为
pb-16S-nt的文件夹。如果是校园网,遇到下载不下来的情况,可以去pb-16S-nt的github主页手动下载,然后上传服务器。 - 在使用
pb-16S分析流程以前,需要安装nextflow和conda,备选singularity或docker。
2. 微生物物种注释分类数据库的下载
$ nextflow run main.nf --download_db - 下载完成以后,当前路径会创建一个名为
databases的文件夹。 - 如果下载不成功可以进行手动下载,下载地址zenodo: https://zenodo.org/records/6912512。
创建
databases的文件夹,将下载文件放入其中。
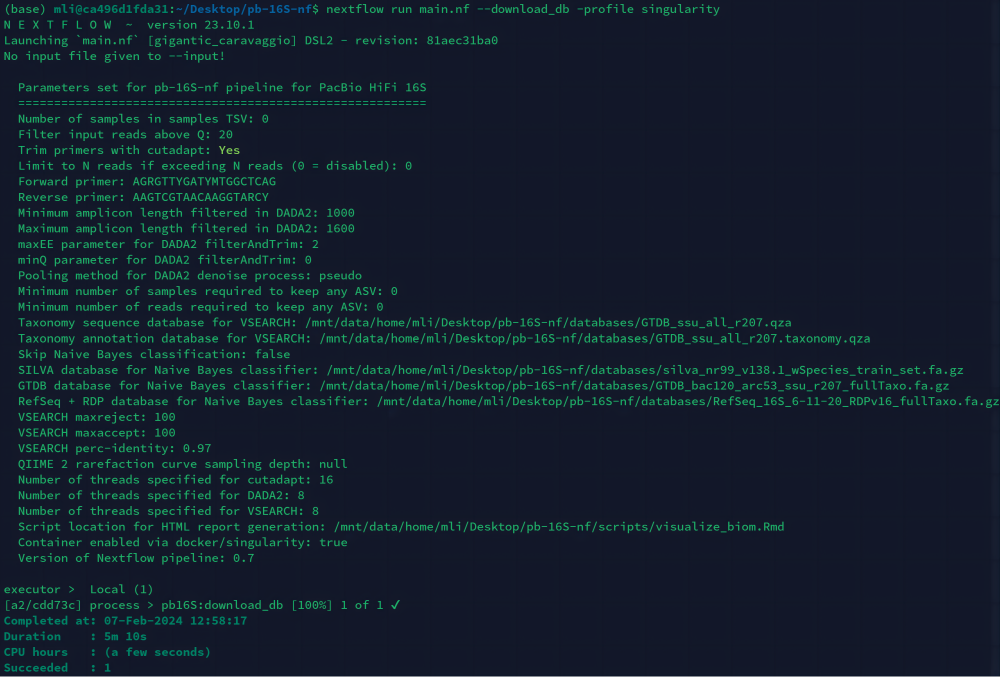
图6. 数据库下载成功示意图
3. 使用示例样本测试软件
# 创建样本TSV文件,用来指定样本路径
$ echo -e "sample-id\tabsolute-filepath\ntest_data\t$(readlink -f test_data/test_1000_reads.fastq.gz)" > test_data/test_sample.tsv
# 测试数据,使用conda创建环境
$ nextflow run main.nf --input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv -profile conda \
--outdir results
# 如果conda创建不了,可以尝试docker或singularity
$ nextflow run main.nf --input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv -profile singularity \
--outdir results- 如果因为网络原因,
conda创建不了环境可以参考我在github上提出的解决方案:https://github.com/PacificBiosciences/HiFi-16S-workflow/issues/2。 - 如果
conda创建环境还是不行,可以尝试-profile docker或-profile singularity。 - 如果使用
docker或singularity, 第一次运行测试样本数据,需要下载镜像,等待时间较长。
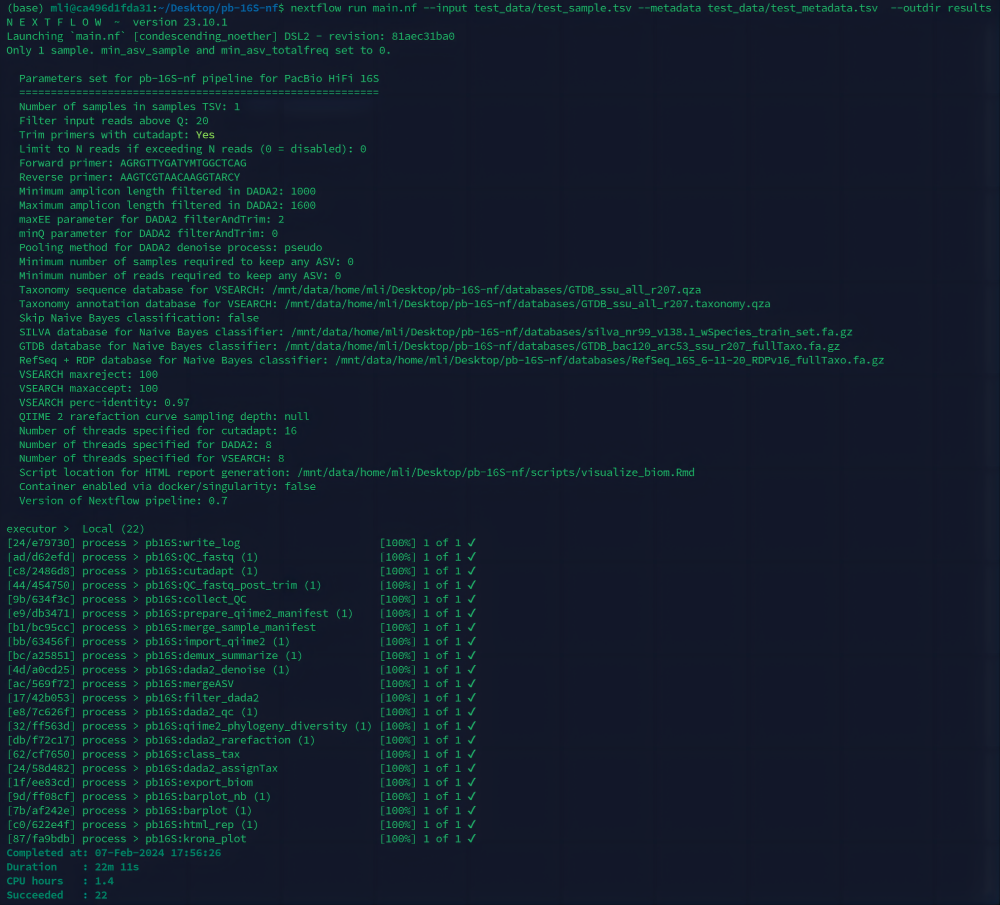
图7. 测试数据运行流程
四、PacBio三代全长16S分析流程
前提是需要安装SMRTlink。
1. 下载 Sequel II 16S barcode序列文件。
在PacBio官网 Multiplexing Page 里下载 barcode 的 Fasta 文件 (图7)。
图7. 全长16S barcode序列fasta文件下
2. 上传文件至服务器,导入SMRTlink中。
- 将
Sequel_16S_barcodes_for_192-Plex.fasta文件上传至服务,放在opt/barcodes/路径下,没有此路径可以自己创建。 - 通过
Data Management - Import Data - Select Barcodes (FASTA)文件导入SMRTlink软件,后面拆分barcode使用(图8)。
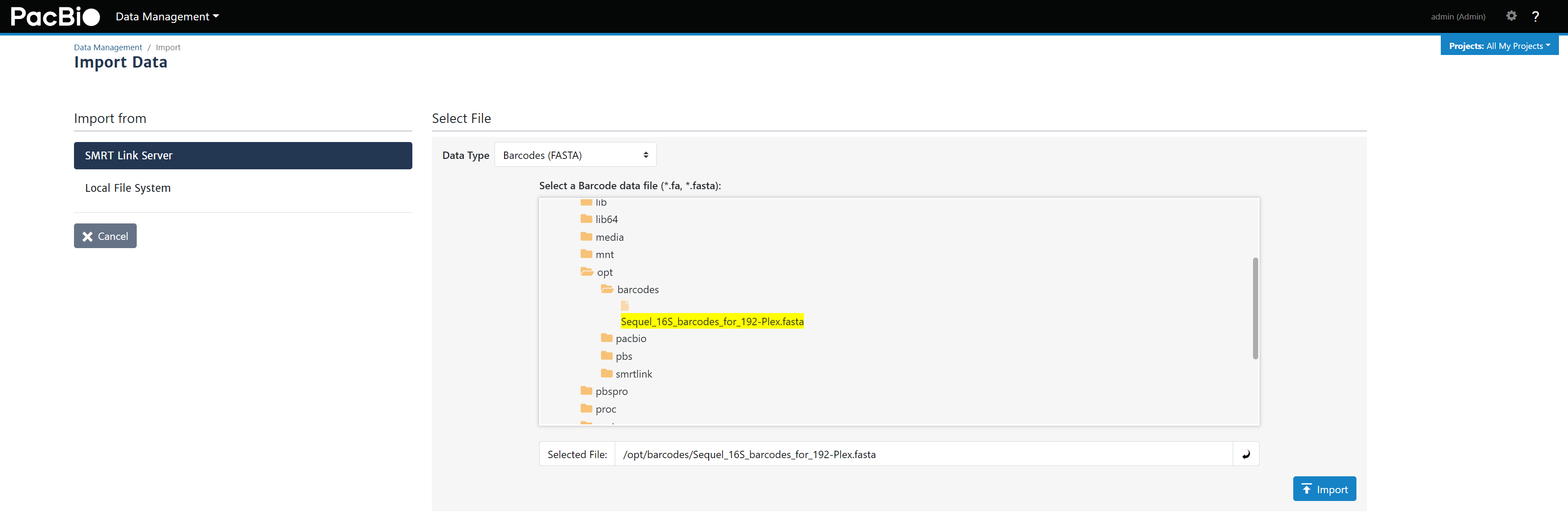
图8. 将fasta文件导入SMRTlink中
3. 原始下机数据跑CCS流程,跑Demultiplex Barcodes流程。
- 原始下机数据跑
CCS流程。 - 需要根据示例制作
Barcoded Sample File,目的是将barcode和样本名称对应起来。 - 通过
Demultiplex Barcodes流程将混样样本(hifi reads)拆分,SMRT Analysis - Creat New Analysis - Demultiplex Barcodes,并按照图9设置。
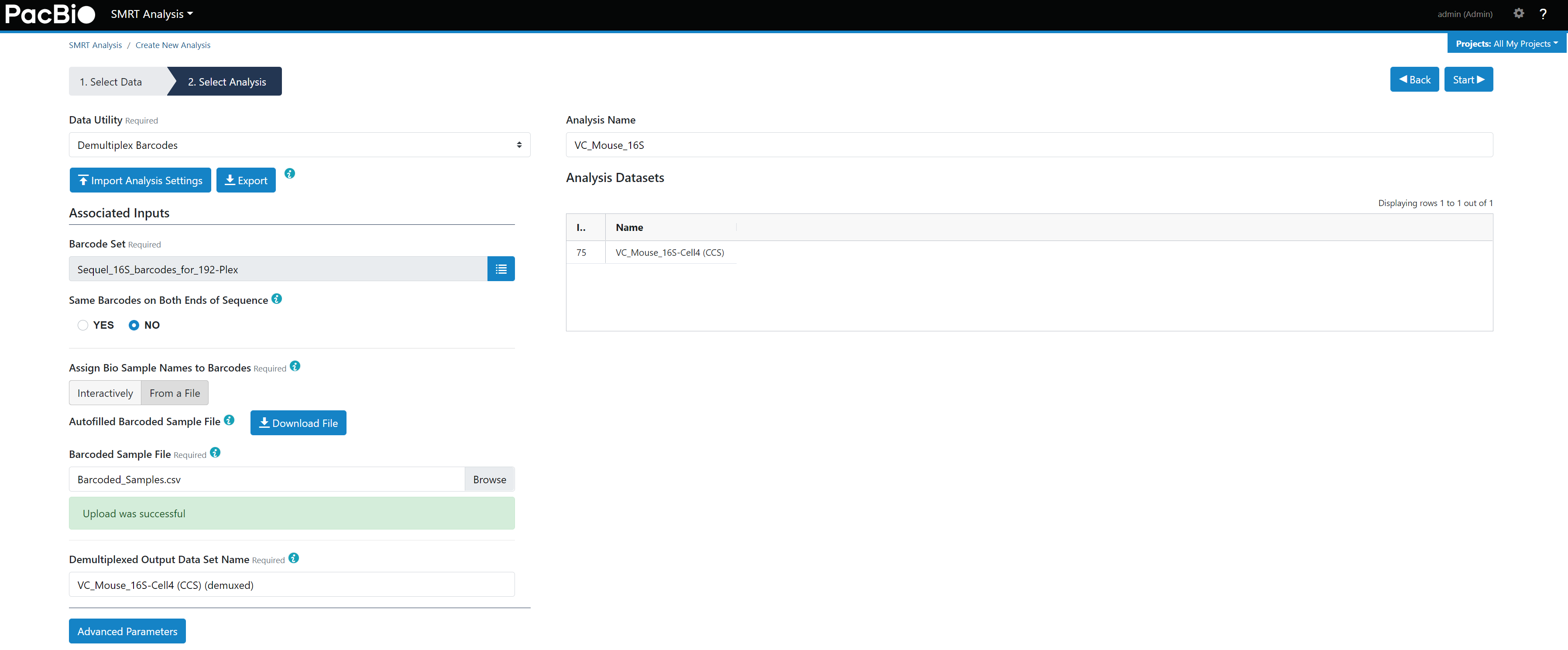
图9. Demultiplex Barcodes流程
4. 文件拷贝及重命名。
- 拆分后的样本以
demultiplex.barcode组合.hifi_reads.fastq.gz命名 (图10)。 - 可以将所有文件下载保存,或上传分析服务器进行后续全长16S分析。
- 可用以下代码,将样本重新命名。
$ cat rename.txt
demultiplex.barcode组合.hifi_reads.fastq.gz newname1.fastq.gz
demultiplex.barcode组合.hifi_reads.fastq.gz newname2.fastq.gz
$ cat rename.txt | while read i j
>do
>mv $i $j
>done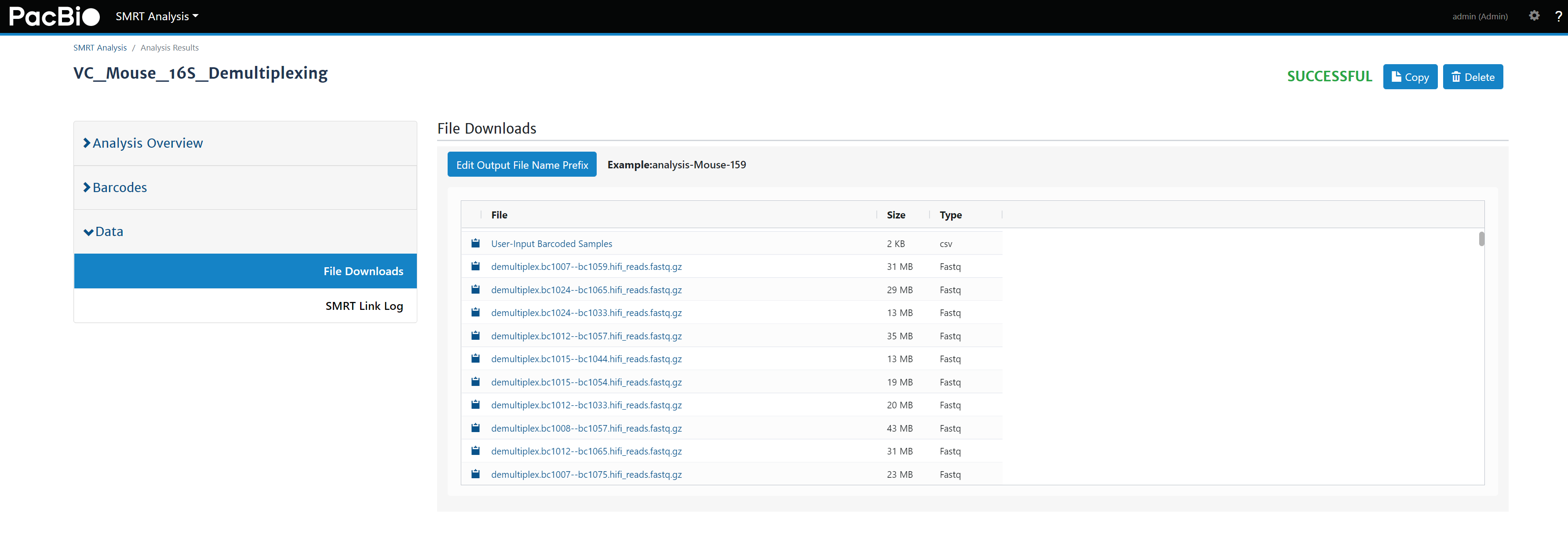
图10. 拆分后的fastq文件
5. 进行pb-16S-nt流程的分析。
根据要求制作metadata.tsv 和 sample.tsv两个文件,就可以按照示例进行PacBio全长16S分析流程了。
6. 运行实际样本
$ nohup nextflow run main.nf --input 16S_project/sample.tsv \
--metadata 16S_project/metadata.tsv -profile conda \
--outdir 16S_project_results &
# 在获得rarefaction曲线后,可以指定rarefaction深度,重新跑程序
$ nohup nextflow run main.nf --input 16S_project/sample.tsv \
--metadata 16S_project/metadata.tsv -profile conda \
--outdir 16S_project_results \
-resume --rarefaction_depth 5000 &7. 结果文件
具体的结果解读可以参照:https://github.com/PacificBiosciences/HiFi-16S-workflow/blob/main/pipeline_overview.md。

图11. HiFi Full-length 16S分析流程结果
P.S:
1.如果没有安装SMRTlink,barcode的拆分也可以使用lima。
#HiFi run from BAM with symmetric barcodes:
$ lima <movie>.hifi_reads.bam barcodes.fasta <movie>.demux.bam --hifi-preset SYMMETRIC2.如果数据来自测序服务商,样本数据应该都是拆分好的,直接使用HiFi Full-length 16S分析流程分析即可。
五、Nextflow软件的安装
Nextflow官网: https://www.nextflow.io/
#确保java11已经安装
$ java -version
#如果没有安装java,运行下面命令进行安装
#安装OpenJDK 11 JDK, centOS7服务器系统
$ yum install java-11-openjdk-devel
#安装nextflow
$ curl -s https://get.nextflow.io | bash
#nextflow 试运行
./nextflow run hello
#可以把nextflow加入到系统路径当中参考文献:
- David M. Ward, Roland Weller, Mary M. Bateson, 16S rRNA sequences reveal uncultured inhabitants of a well-studied thermal community, FEMS Microbiology Reviews,1990。
- 三代全长16s — 望向微生物世界的尽头。
- Matsuo, Y., Komiya, S., Yasumizu, Y. et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol 21, 35 (2021)。
- PacBio 16S全长测序:一种高效且经济的微生物组研究方法 。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号