sra转fastq
原创

因为恰好遇到了PRJNA752099这个数据集,他上传的fastq文件被合并成了一个,所以我需要下载SRA文件重新拆分。正好作为上游最后一块的补充内容。
下载sratoolkit
wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
tar -vxzf sratoolkit.tar.gz
export PATH=$PATH:$PWD/sratoolkit.3.1.1-ubuntu64/bin
faster-dump --help
我事先下载了PRJNA752099的4个SRA文件,所以这里从转fastq开始。
mkdir fastq_store
ls SRR* > list.txt
#sh脚本
samples=$(<list.txt)
for s in ${samples};do
echo "${s} starts at $(date)"
fasterq-dump ./${s} -O fastq_store -e 10 --include-technical -S
echo "${s} finished at $(date)"
done这里解释一下faster-dump需要设置--include-technical才能读取barcode之类的reads,而且无法输出.gz格式的压缩文件,需要自行压缩。
运行完成后可以看到每个样本输出了3个fastq文件。

分别查看这几个文件,可以看到1是索引信息,2中是barcode,3是对应R2的长读
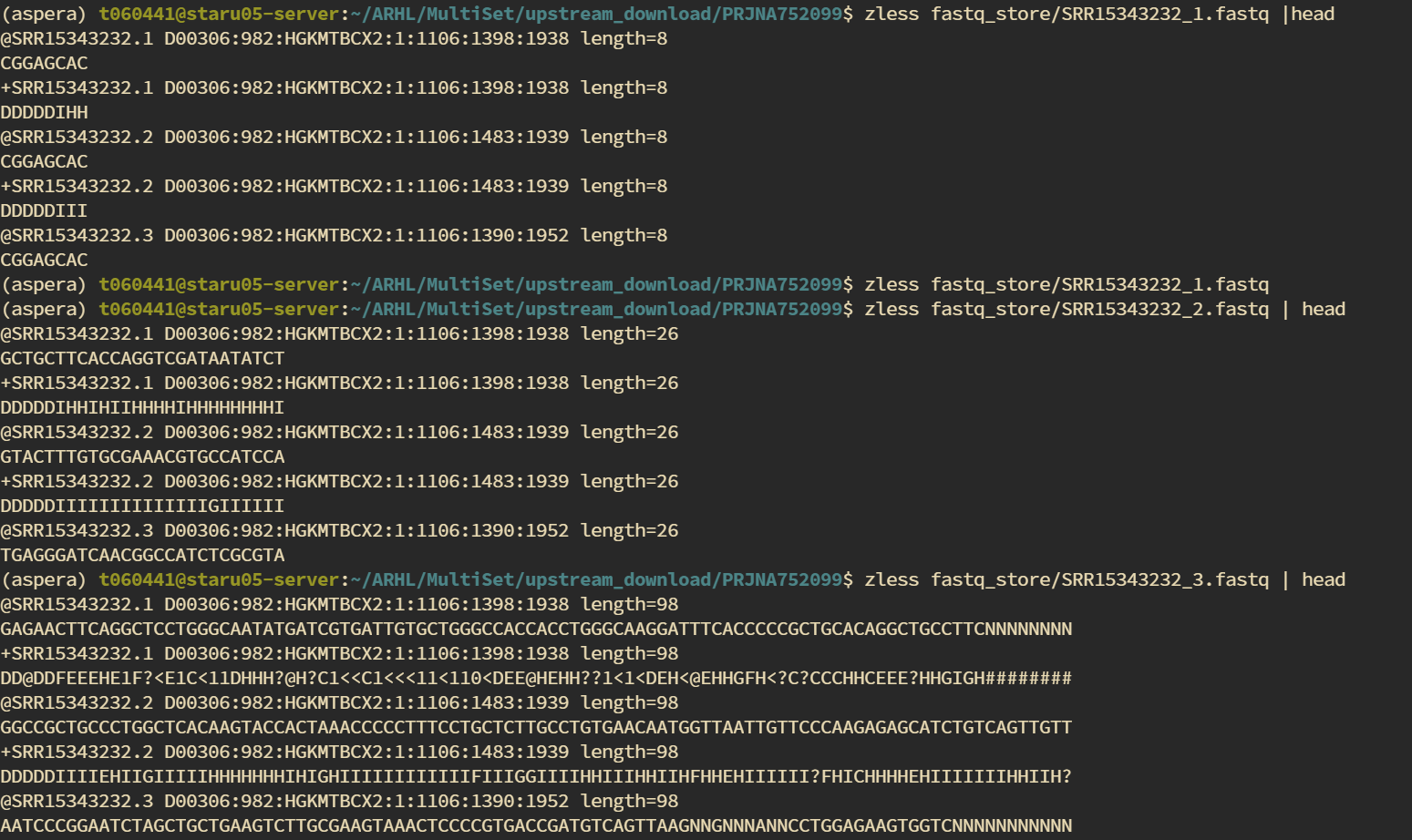
接下来只需要压缩后软链接到cellranger脚本路径下改名然后开始定量啦。
nohup gzip SRR*_1.fastq SRR*_2.fastq SRR*_3.fastq &原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号