Visium HD数据分析之空间聚类算法Banksy
原创
Visium HD数据分析之空间聚类算法Banksy
原创
生信大杂烩
发布于 2025-05-29 21:38:11
发布于 2025-05-29 21:38:11
前面我们通过Stardist或Cellpose完成了细胞核/细胞分割,然后将HD的2um bin以各种方法进行聚合,得到了接近snRNA或scRNA数据,接下来我们就可以对这些数据进行降维聚类,进行后续分析。但是传统的单细胞聚类算法,例如Seurat和Scanpy中的lovain和leiden等聚类算法,通常在处理空间数据时忽略了空间信息。然而,由于细胞状态受其周围细胞的影响,将转录组数据与细胞的空间信息结合起来进行聚类分析,将更有助于揭示细胞在组织中的分布和相互作用。
2024年2月,Nature Genetics上发表了一篇题为BANKSY unifies cell typing and tissue domain segmentation for scalable spatial omics data analysis(https://www.nature.com/articles/s41588-024-01664-3)的研究文章。这项工作开发了一种名为BANKSY的空间聚类算法。BANKSY可通过结合细胞/Spots的转录组信息和其局部微环境信息,显著提升空间组学数据分析的准确性和效率。BANKSY算法不仅能够处理大规模数据集,提高细胞类型聚类和组织域分割的性能,还展现了在多种RNA和蛋白质数据集上的广泛适用性。
具体来说,BANKSY是一种聚类空间组学数据的方法,通过将每个细胞的特征与其空间近邻特征的平均值以及邻域特征梯度相结合来增强。通过将邻域信息纳入聚类中,BANKSY能够:
在嘈杂的数据中改善细胞/Spots类型分配;
区分微环境分层的微妙不同细胞/Spots类型;
确定共享相同微环境的空间域。
BANKSY官方教程详见:
R语言版本:https://prabhakarlab.github.io/Banksy/
Python版本:https://github.com/prabhakarlab/Banksy_py
一. BANKSY算法的工作流程
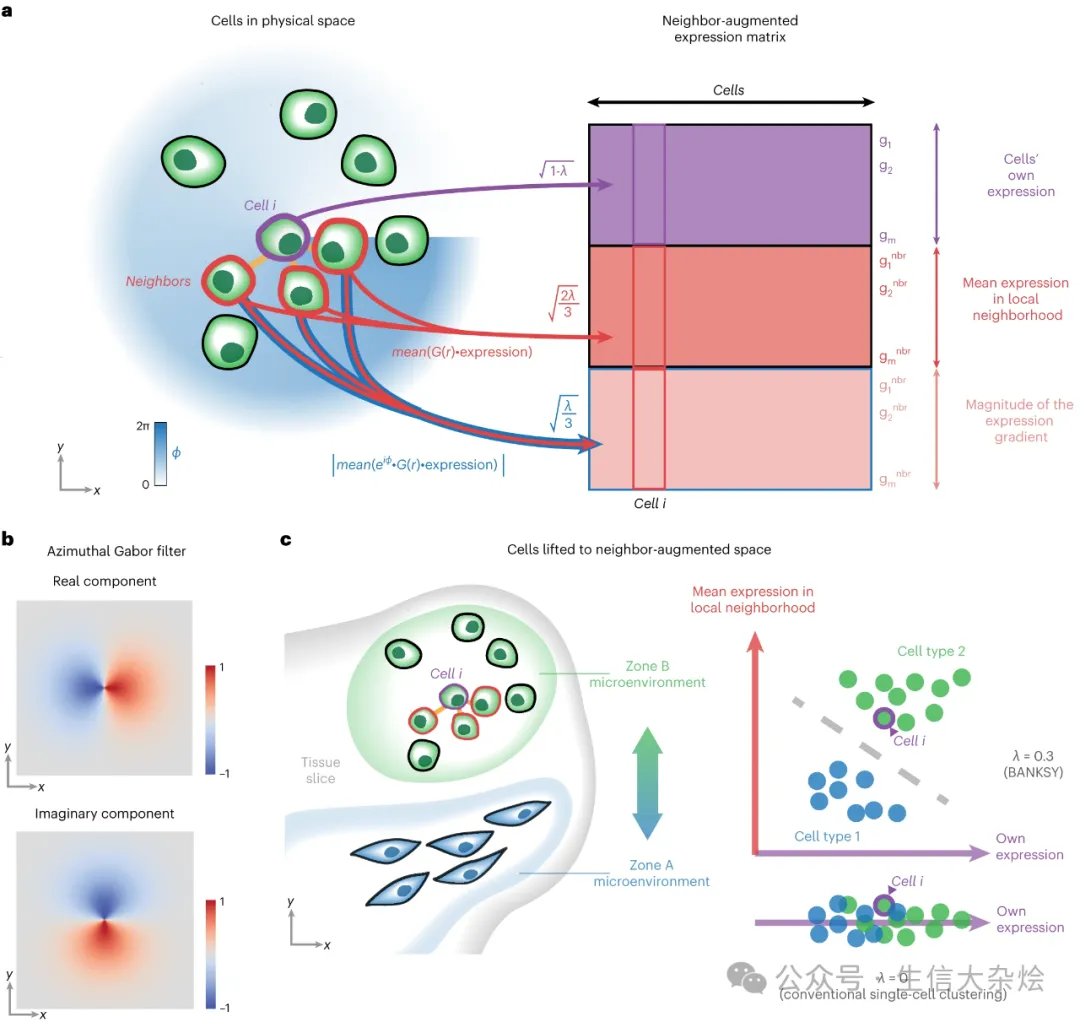
图片
一种空间聚类的策略是将细胞的空间坐标附加到它们的基因表达向量中。然而,这会导致空间上相距较远的细胞分入不同的聚类中,即使它们具有相同的转录组。一种更直观的方法是附加某种表示细胞微环境的表示。BANKSY算法采用平均邻域表达和AGF(图1a,1b)来代表每个细胞周围的转录组微环境。重要的是,AGF(图1b)可以被视为衡量每个细胞邻域内基因表达梯度,对样本旋转是不变的。接下来,这些额外的特征被用来将细胞嵌入到一个邻域增强的乘积空间中(图1c)。经过降维处理,然后在结果嵌入空间中构建图形,可以使用任何图划分算法进行聚类。默认情况下,BANKSY使用Leiden社区检测算法,因为其速度和可扩展性,虽然也提供了其他方法(Louvain、基于模型的聚类和k-means)作为选项。
为了控制细胞自身和邻域特征对嵌入中细胞-细胞距离的相对贡献,BANKSY使用一个混合参数,λ∈ [0, 1],来权衡细胞转录组矩阵和邻域表达矩阵(平均值和AGF;图1)的贡献。较小的λ设置强调细胞自身的转录组,因此导致细胞根据细胞类型进行聚类。在极限情况下,当λ = 0时,BANKSY简化为传统的非空间聚类。通过增加λ,增加邻域签名的影响,可以将BANKSY从细胞类型聚类模式切换到组织域分割模式,导致细胞根据组织域进行聚类。
二. BANKSY的python代码实现
import osimport numpy as npimport pandas as pdimport scanpy as scfrom banksy.main import median_dist_to_nearest_neighbourfrom banksy.initialize_banksy import initialize_banksyfrom banksy.embed_banksy import generate_banksy_matrixfrom banksy.main import concatenate_allfrom banksy_utils.umap_pca import pca_umapfrom banksy.cluster_methods import run_Leiden_partitionfrom banksy.plot_banksy import plot_resultsimport matplotlib.pyplot as pltimport warningswarnings.filterwarnings("ignore")plt.style.use('default')plt.rcParams['figure.facecolor'] = 'white'
adata = sc.read_h5ad('./adata_cellpose.h5ad')adata.var_names_make_unique()adata.obs['sample'] = 'P1_CRC'
# 质控adata.var['mt'] = adata.var_names.str.upper().str.startswith('MT-') adata.var['ribo'] = adata.var_names.str.upper().str.startswith(("RPS","RPL"))sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)adata.obs['log10GenesPerUMI'] = np.log10(adata.obs['n_genes_by_counts']) / np.log10(adata.obs['total_counts'])
# 细胞过滤sc.pp.filter_cells(adata, min_counts=30)sc.pp.filter_genes(adata, min_cells=10)adata = adata[(adata.obs['log10GenesPerUMI'] > 0.8) & (adata.obs['log10GenesPerUMI'] < 0.99) & (adata.obs['pct_counts_mt'] < 20)]
# Normalizeadata.layers["counts"] = adata.X.copy()sc.pp.normalize_total(adata)
# HVGsc.pp.log1p(adata)sc.pp.highly_variable_genes( adata, flavor='seurat', n_top_genes=2000)
## 构建空间最近邻图# Find median distance to closest neighboursnbrs = median_dist_to_nearest_neighbour(adata, key="spatial")
## 生成距离空间权重(Generate spatial weights from distance)k_geom = 15 # number of spatial neighboursmax_m = 1 # use both mean and AFTnbr_weight_decay = "scaled_gaussian" # can also choose "reciprocal", "uniform" or "ranked"coord_keys = ('xcoord', 'ycoord', 'spatial')banksy_dict = initialize_banksy( adata, coord_keys, k_geom, nbr_weight_decay=nbr_weight_decay, max_m=max_m, plt_edge_hist=True, # Visualize the edge histogram to show the histogram of distances between cells and the weights plt_nbr_weights=True, # Visualize weights by plotting the connections. plt_agf_angles=False, # Visualize weights with Azimuthal angles plt_theta=True, # Visualize angles around random cell. Plot points around a random index cell, annotated with angles from the index cell.)
## 生成BANKSY矩阵# BANKSY几个主要超参数resolutions = [0.5] # clustering resolution for UMAPpca_dims = [20] # Dimensionality in which PCA reduces tolambda_list = [0.2] # list of lambda parametersbanksy_dict, banksy_matrix = generate_banksy_matrix( adata, banksy_dict, lambda_list, max_m )
## 将非空间结果附加到banksy_dict以进行比较(不必要)banksy_dict["nonspatial"] = { # Here we simply append the nonspatial matrix (adata.X) to obtain the nonspatial clustering results 0.0: {"adata": concatenate_all([adata.X], 0, adata=adata),}}
## PCA降维, UMAP可视化pca_umap( banksy_dict, pca_dims = pca_dims, # 这里PCA降到20个维度 add_umap = True, plt_remaining_var = False,)
## 细胞聚类--默认情况下,建议使用基于分辨率的聚类(即leiden 或 louvain)results_df, max_num_labels = run_Leiden_partition( banksy_dict, resolutions, num_nn = 50, # k_expr: number of neighbours in expression (BANKSY embedding or non-spatial) space num_iterations = -1, # run to convergenece partition_seed = 12345, match_labels = True,)results_df中包含了空间聚类的结果,使用results_df['adata']['scaled_gaussian_pc20_nc0.20_r0.60']就可以获取BANKSY降维聚类结果。
from banksy.plot_banksy import plot_resultsc_map = 'tab20' # specify color mapweights_graph = banksy_dict['scaled_gaussian']['weights'][0]plot_results( results_df, weights_graph, c_map, match_labels = True, coord_keys = coord_keys, max_num_labels = max_num_labels, save_path = 'tmp_png', save_fig = True)BANKSY考虑空间位置聚类结果展示
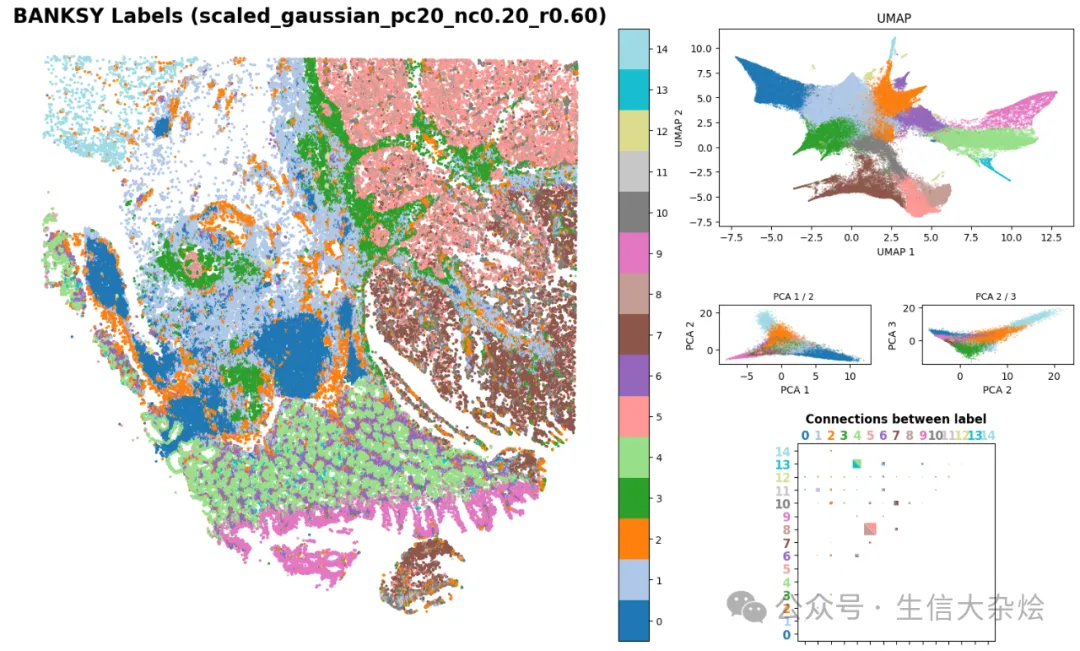
图片
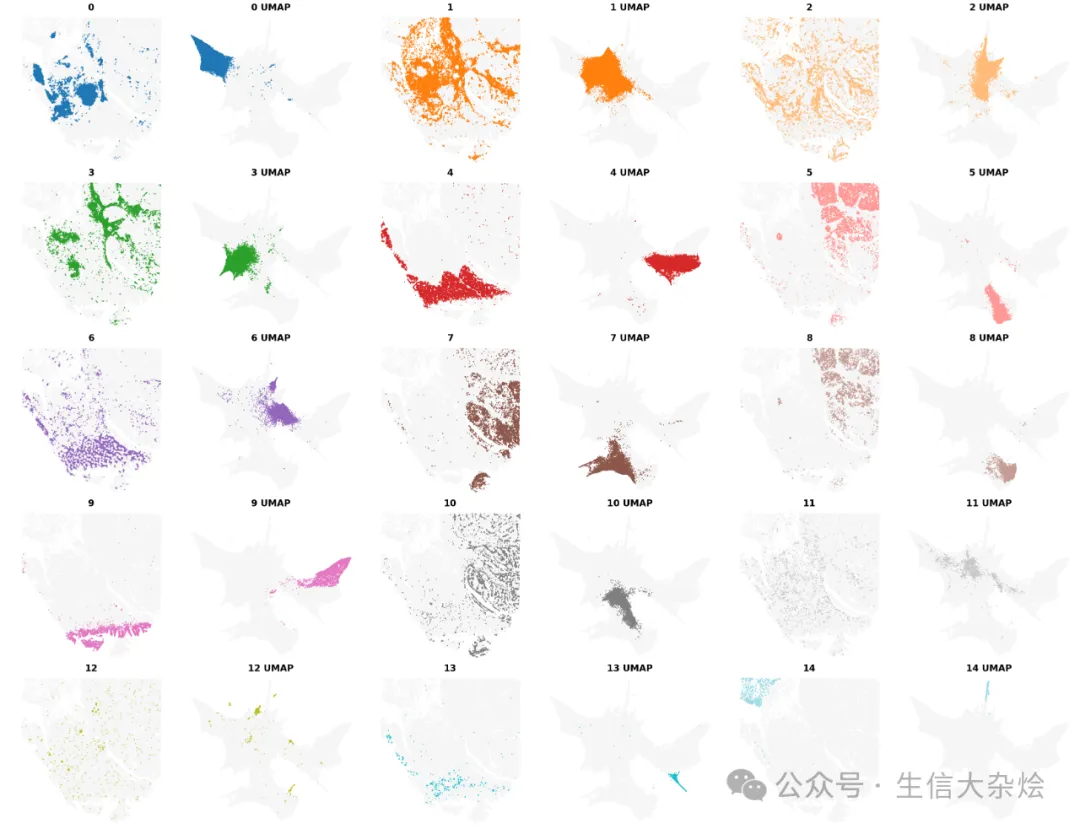
图片
传统单细胞降维聚类结果展示
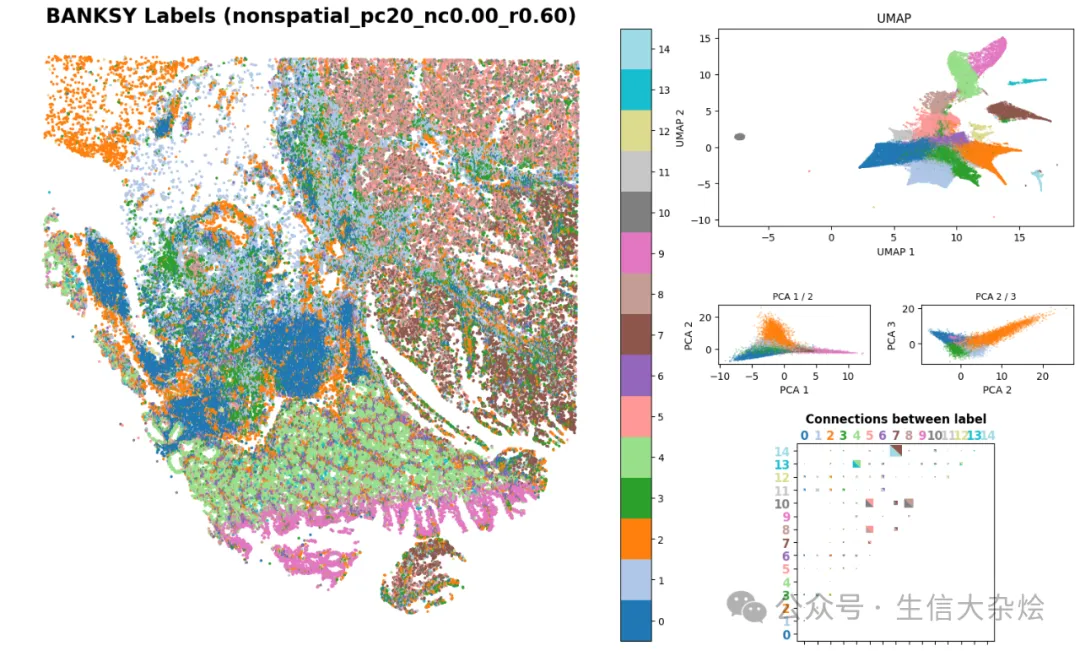
图片
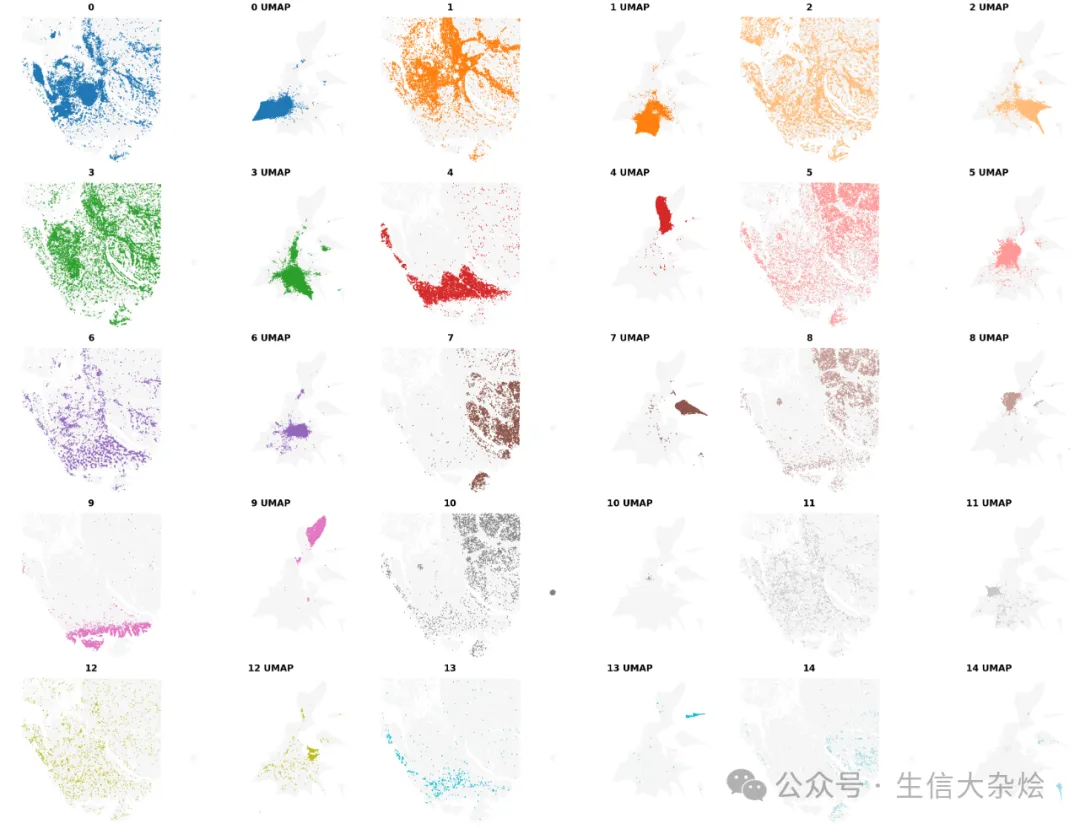
图片
总结
相比于传统的单细胞聚类算法,BANKSY在细胞/Spots的表达矩阵的基础上,进一步联合了空间坐标信息,这种策略能够有效提高细胞/Spots分类的准确性和效率,进而揭示细胞间的相互作用和微环境影响。此外,BANKSY在处理大规模数据集时运行速度非常快,且兼容多样本分析以及传统的整合分析算法如Harmony。最后,BANKSY适用于多种空间数据类型(例如 10x Visium、Slide-seq、MERFISH、CosMX、CODEX、Visium HD)。
题外话
这里我们演示的是一个HD样本空间聚类结果,如果有多个样本,只需要对每个样本进行单独处理后,然后将多个样本的adata进行concat后再harmony或者bbknn合并即可。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号