从零开始的异世界生信学习 GEO数据库数据挖掘--GEO代码-芯片数据分析-1
原创
从零开始的异世界生信学习 GEO数据库数据挖掘--GEO代码-芯片数据分析-1
原创
用户10361520
修改于 2023-03-09 15:33:10
修改于 2023-03-09 15:33:10
生信技能树
1.代码相关R包的加载
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")
if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
cran_packages <- c('tidyr',
'tibble',
'dplyr',
'stringr',
'ggplot2',
'ggpubr',
'factoextra',
'FactoMineR',
'devtools',
'cowplot',
'patchwork',
'basetheme',
'paletteer',
'AnnoProbe',
'ggthemes',
'VennDiagram',
'tinyarray')
Biocductor_packages <- c('GEOquery',
'hgu133plus2.db',
'ggnewscale',
"limma",
"impute",
"GSEABase",
"GSVA",
"clusterProfiler",
"org.Hs.eg.db",
"preprocessCore",
"enrichplot")
for (pkg in cran_packages){
if (! require(pkg,character.only=T) ) {
install.packages(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
for (pkg in Biocductor_packages){
if (! require(pkg,character.only=T) ) {
BiocManager::install(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
for (pkg in c(Biocductor_packages,cran_packages)){
require(pkg,character.only=T)
}2.芯片表达矩阵数据下载
#清空环境,加载R包
rm(list = ls())
library(GEOquery)
#下载GEO数据先去GEO数据库网页确定是否是表达芯片数据,不是的话不能用本流程。
gse_number = "GSE56649"
eSet <- getGEO(gse_number, destdir = '.', getGPL = F)
##getGEO函数可以下载到工作目录下和读取GSE文件,
class(eSet)
length(eSet)
eSet = eSet[[1]]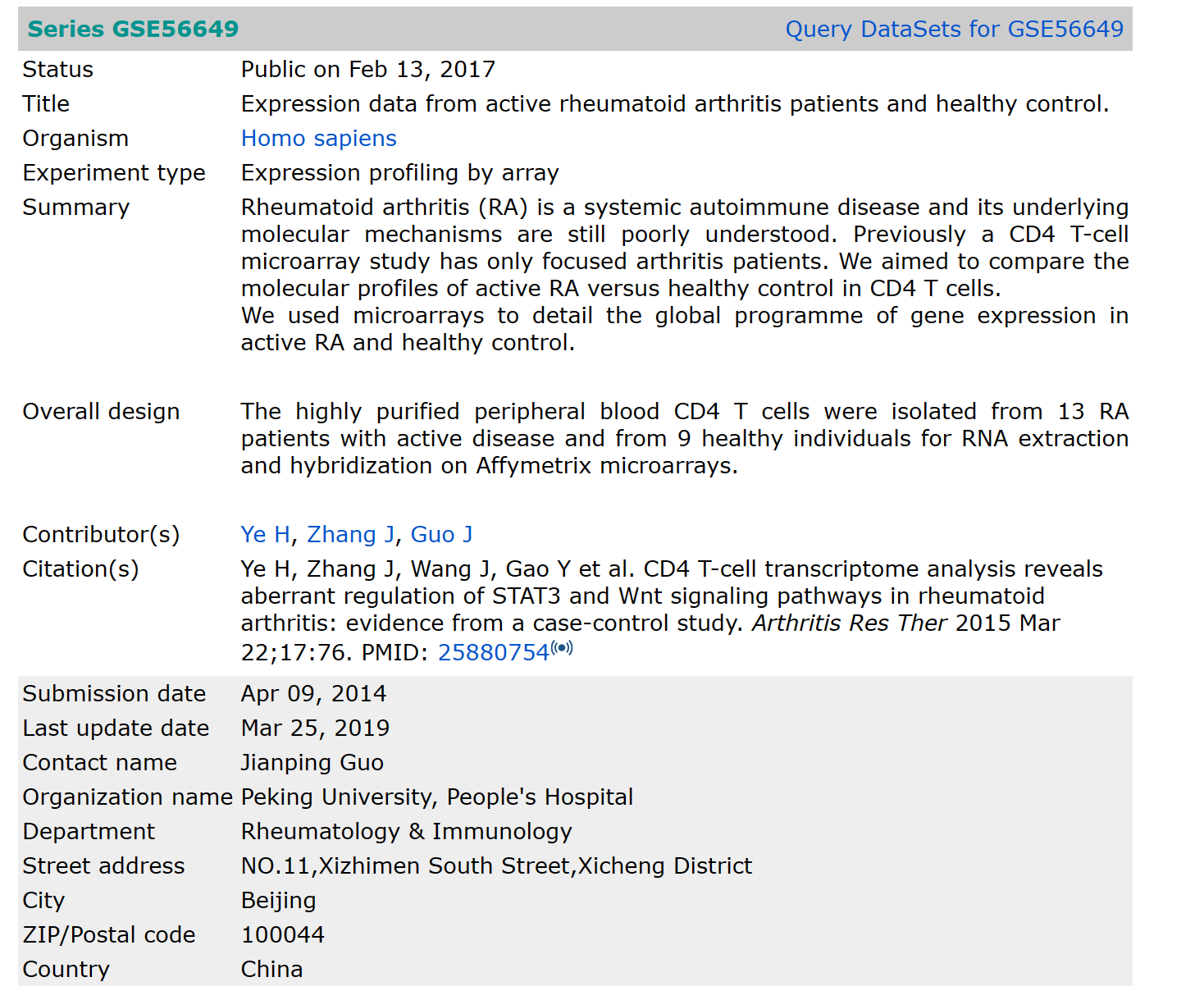
网页查看表达矩阵数据
在GEO数据库网页中可以查看数据的基本信息
注意!!!array芯片数据才可以用此代码分析
GEO文件下载并读取到R中为只有一个元素的list
在列表中取子集后得到"ExpressionSet"结构数据,为"Biobase"包中的数据形式
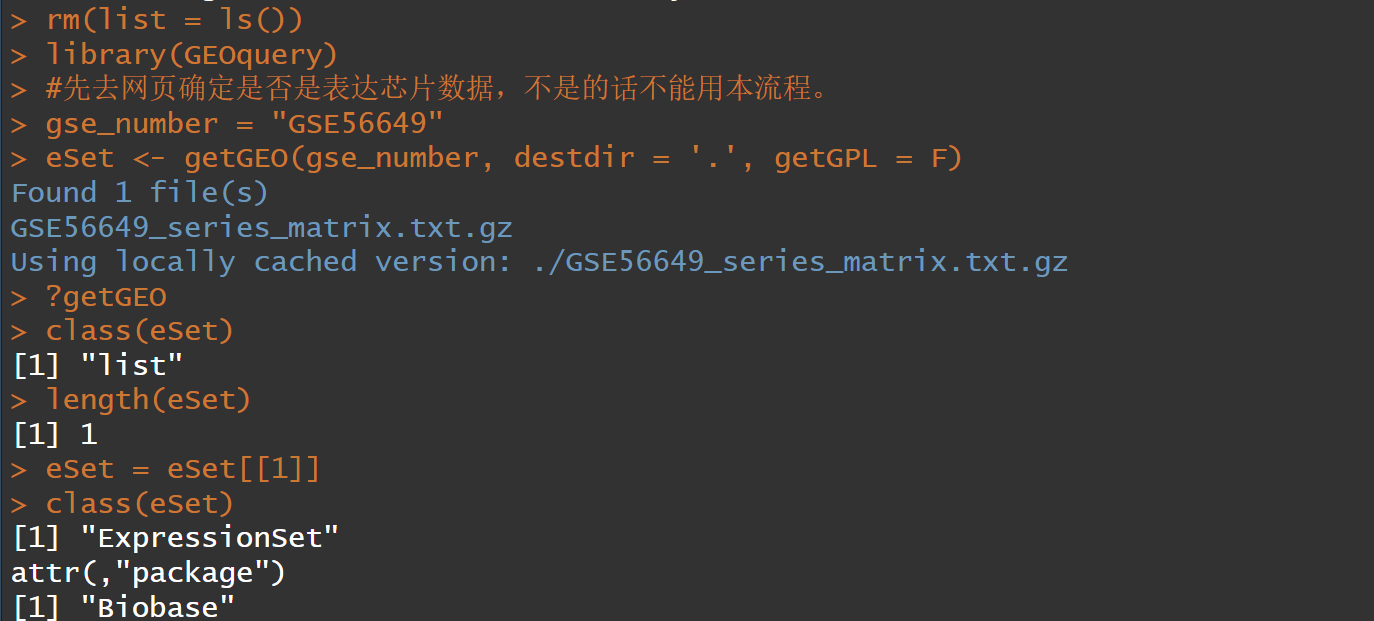
#(1)提取表达矩阵exp
exp <- exprs(eSet)
##exprs函数是expressionset文件中提取表达矩阵的函数
dim(exp)
exp[1:4,1:4]
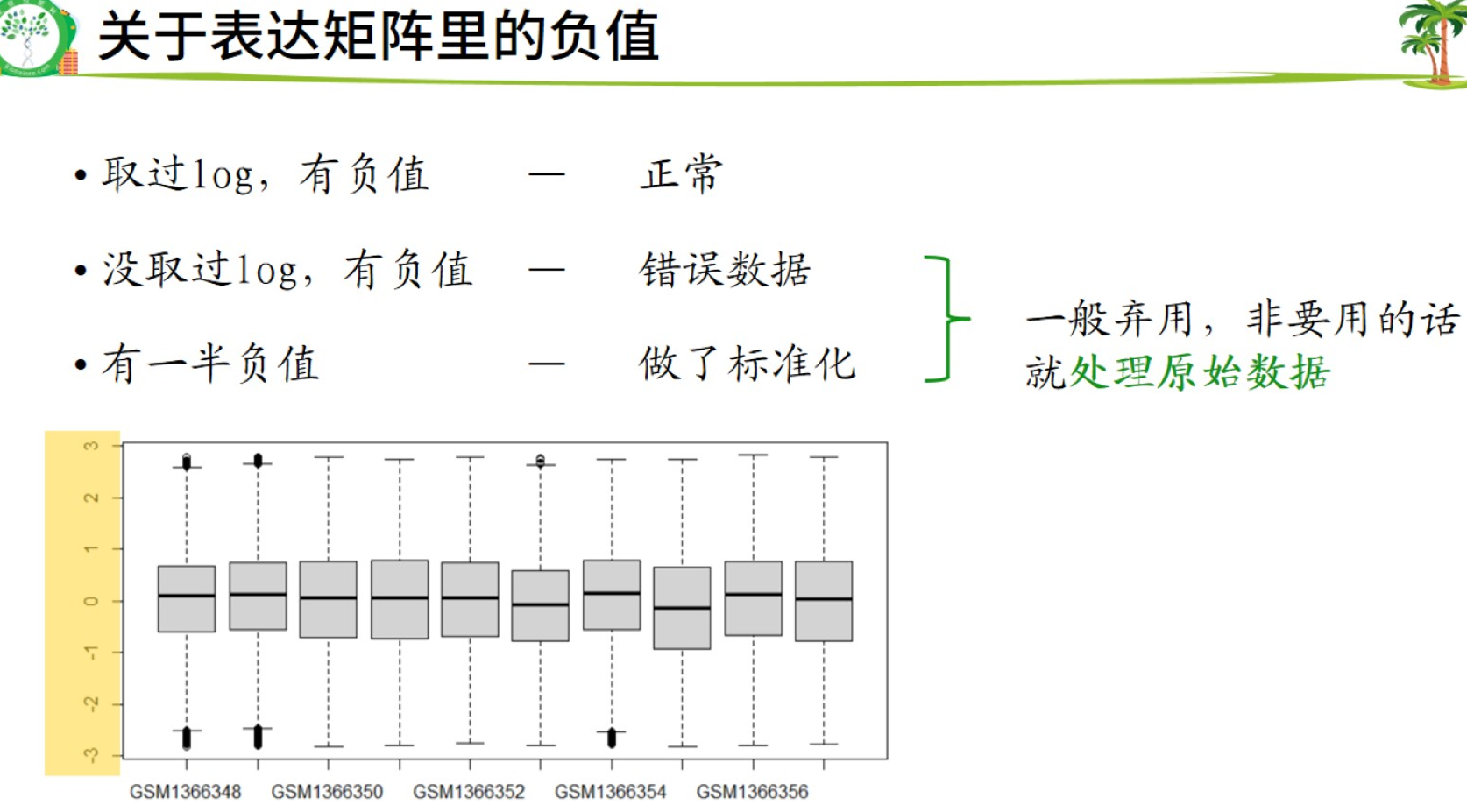
#检查矩阵是否正常,如果是空的就会报错,空的和有负值的、有异常值的矩阵需要处理原始数据。
#如果表达矩阵为空,大多数是转录组数据,不能用这个流程(后面另讲)。
#自行判断是否需要log
exp = log2(exp+1)
## 这个步骤在表达矩阵中表达量+1为了防止数据中有0,取log2后出现负无穷
boxplot(exp)
##exp = limma::normalizeBetweenArrays(exp) 可以通过这句代码进行对表达矩阵处理
#(2)提取临床信息
pd <- pData(eSet)
##表达矩阵的列名和临床信息的行名必须一致才能进行后续分析操作
#(3)让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) exp = exp[,match(rownames(pd),colnames(exp))]
#(4)提取芯片平台编号
gpl_number <- eSet@annotation;gpl_number ##狭义的数据类型取子集需要用@或者$符号,可以进行实验能否补齐
save(gse_number,pd,exp,gpl_number,file = "step1output.Rdata")
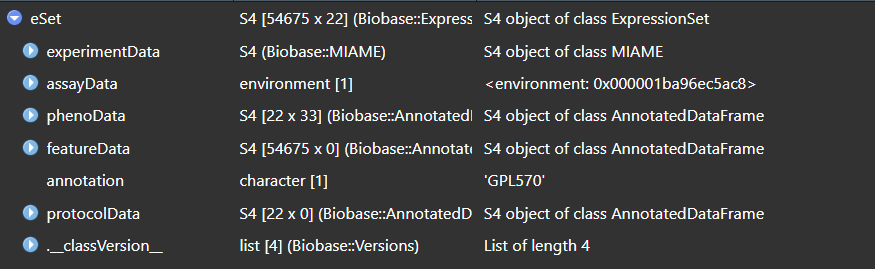
ExpressionSet数据
assayData : 表达矩阵
phenoData : 临床信息
annotation : 芯片型号编号GPL编号
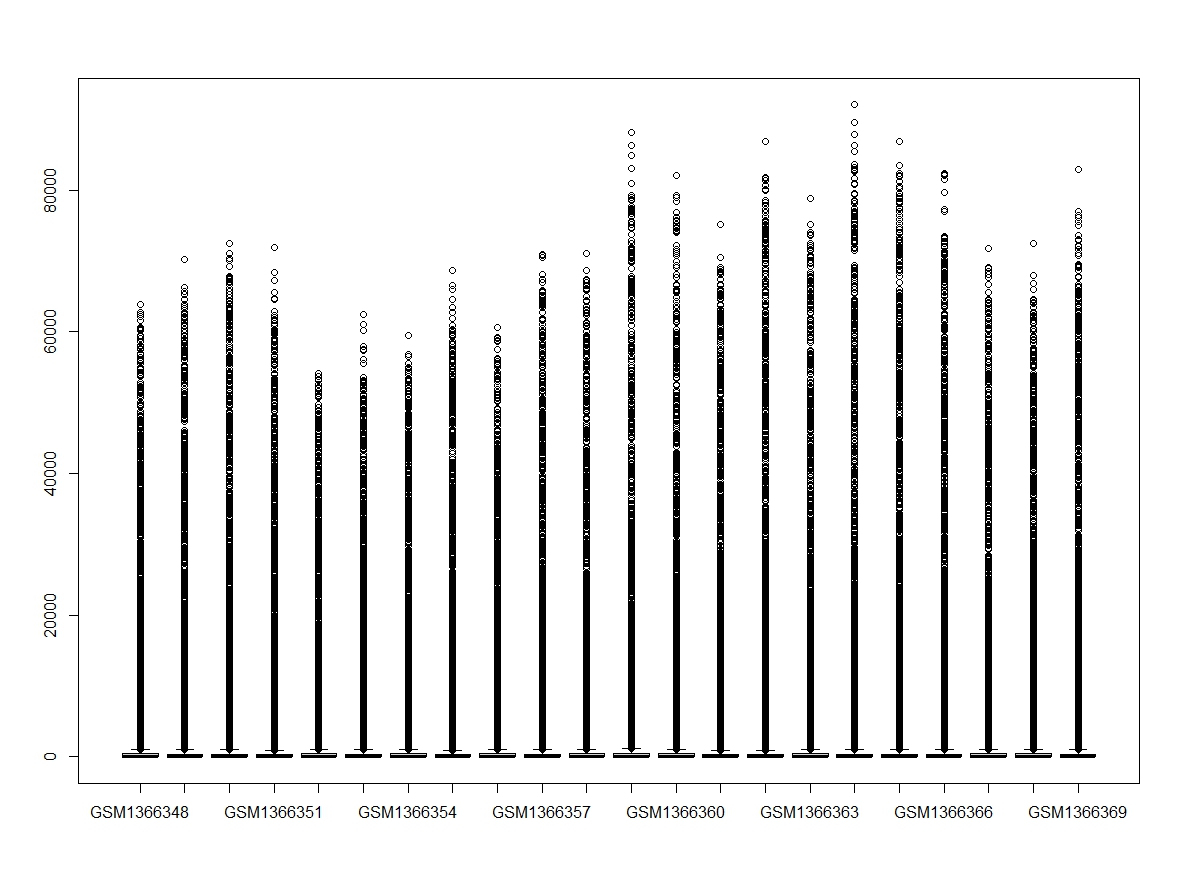
表达量未取log2的箱线图
根据表达矩阵的数值判断是否需要取log2,一般log2的值在0-20左右。
判断表达矩阵是否正常可以绘制箱线图
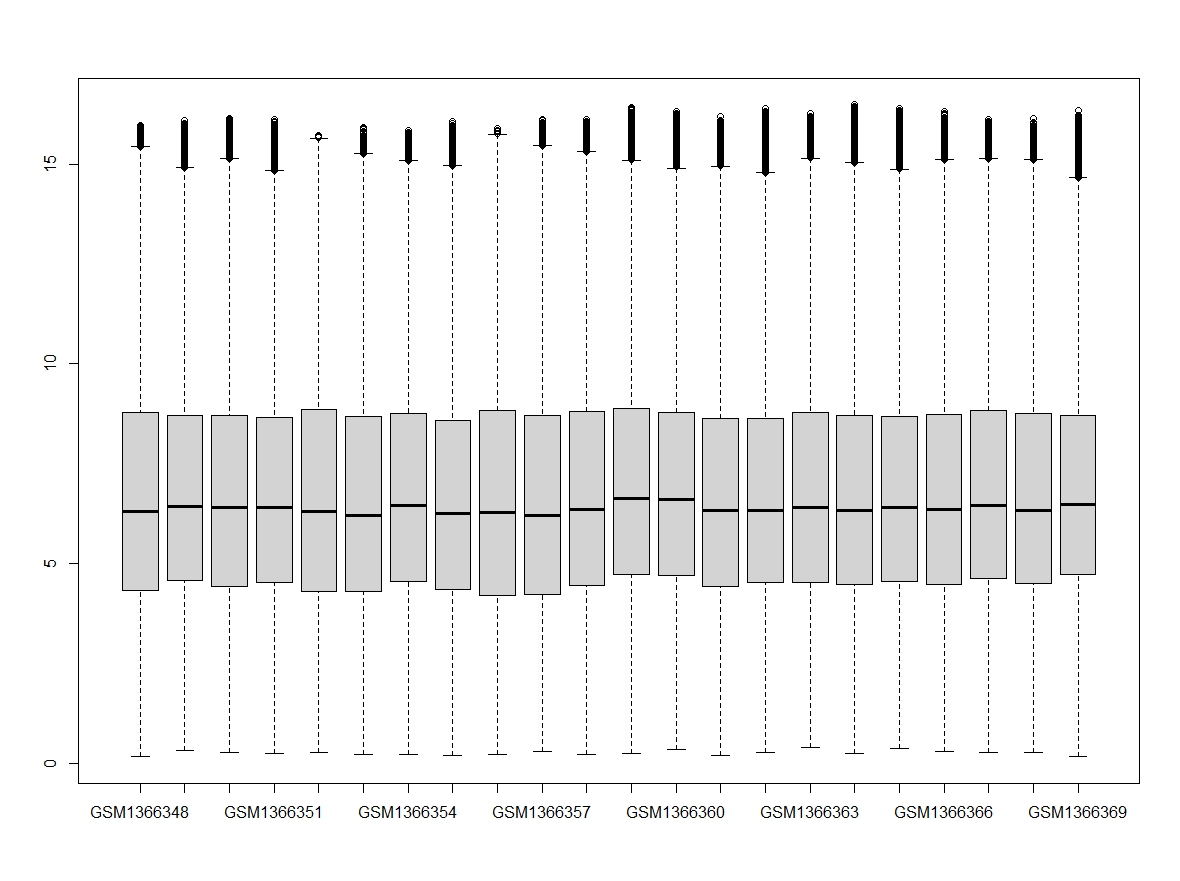
表达量取log2后的箱线图
正常的表达矩阵的箱线图中,中位数线以及上下四分位数线基本平齐
表达量未取log2的箱线图
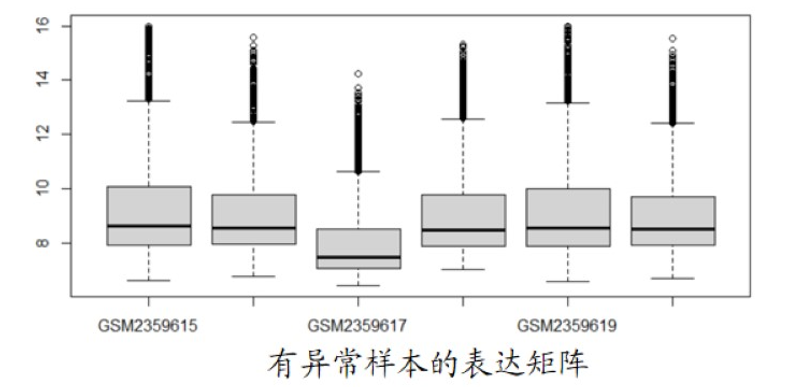
异常表达量的箱线图
图中的GSM2359617为异常样本,其整体基因表达量较其他组低。

rm(list = ls())
library(AnnoProbe)
library(GEOquery)
gse_number = "GSE56649"
eSet = geoChina(gse_number)使用生信技能树服务器快速下载GEO数据,下载为Rdata文件
感谢曾老师!!!以及曾老师的2000元钱!!!
3. 数据实验分组与探针注释
3.1 设置数据的实验分组
设置实验分组的第一步,是根据表格中的数据寻找分组依据。简化关键词,简化为一个单词。
# Group(实验分组)和ids(探针注释)
rm(list = ls())
load(file = "step1output.Rdata")
library(stringr)
# 标准流程代码是二分组,多分组数据的分析后面另讲
# 生成Group向量的三种常规方法,三选一,选谁就把第几个逻辑值写成T,另外两个为F。如果三种办法都不适用,可以继续往后写else if
if(F){
# 1.Group----一般实验分组为一个单词
# 第一种方法,有现成的可以用来分组的列
Group = pd$`disease state:ch1`
## pd$后tab补齐,R语言中,列名存在特殊符号,列名会用反引号标注
## 这种方法适用于临床信息列中分组信息明确
}else if(F){
# 第二种方法,自己生成
Group = c(rep("RA",times=13),
rep("control",times=9))
Group = rep(c("RA","control"),times = c(13,9))
}else if(T){
# 第三种方法,使用字符串处理的函数获取分组
Group=ifelse(str_detect(pd$source_name_ch1,"control"),
"control",
"RA")
##Group=ifelse(str_detect(pd$source_name_ch1,"patient"),"RA", "control")
}
## str_detect用来搜索关键词
##第三种方法需要临床信息中的字符串中有分组信息的文字3.2 将分组数据转变成因子
变量可以分为名义型,有序型或连续性变量。
名义型变量没有顺序之分,比如糖尿病的分类,I型和II型,两者之间没有程度强弱,顺序先后之分,互相独立。
有序性变量表达一种元素间存在顺序之分,但非具体数量关系,例如疾病的病情status(poor,improved,excellent),三者存在程度强弱的关系,poor(较差)不如improved(改善)的病人
连续性变量:可以呈现某个范围之内的任意值。同时表达了数量和顺序。比如年龄age。
因子:在R语言中类别变量(名义型)以及有序类别(有序性)变量称为因子。
# 需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = factor(Group,levels = c("control","RA"))
Group
## factor(Group)生成因子是默认按照首字母顺序排序
##Group = factor(Group,levels = c("control","RA")) 按照代码中的顺序进行排序,control组在第一个位置上
levels:水平 因子里面的取值,顺序十分重要,第一个位置上的是参考水平,为其他取值的对照。因此对照组应该在前,处理组在后,保证差异分析结果不是反的。
3.3 探针注释
获取探针名称和基因注释(gene symbol)对应关系,根据GPL编号获取对应关系。
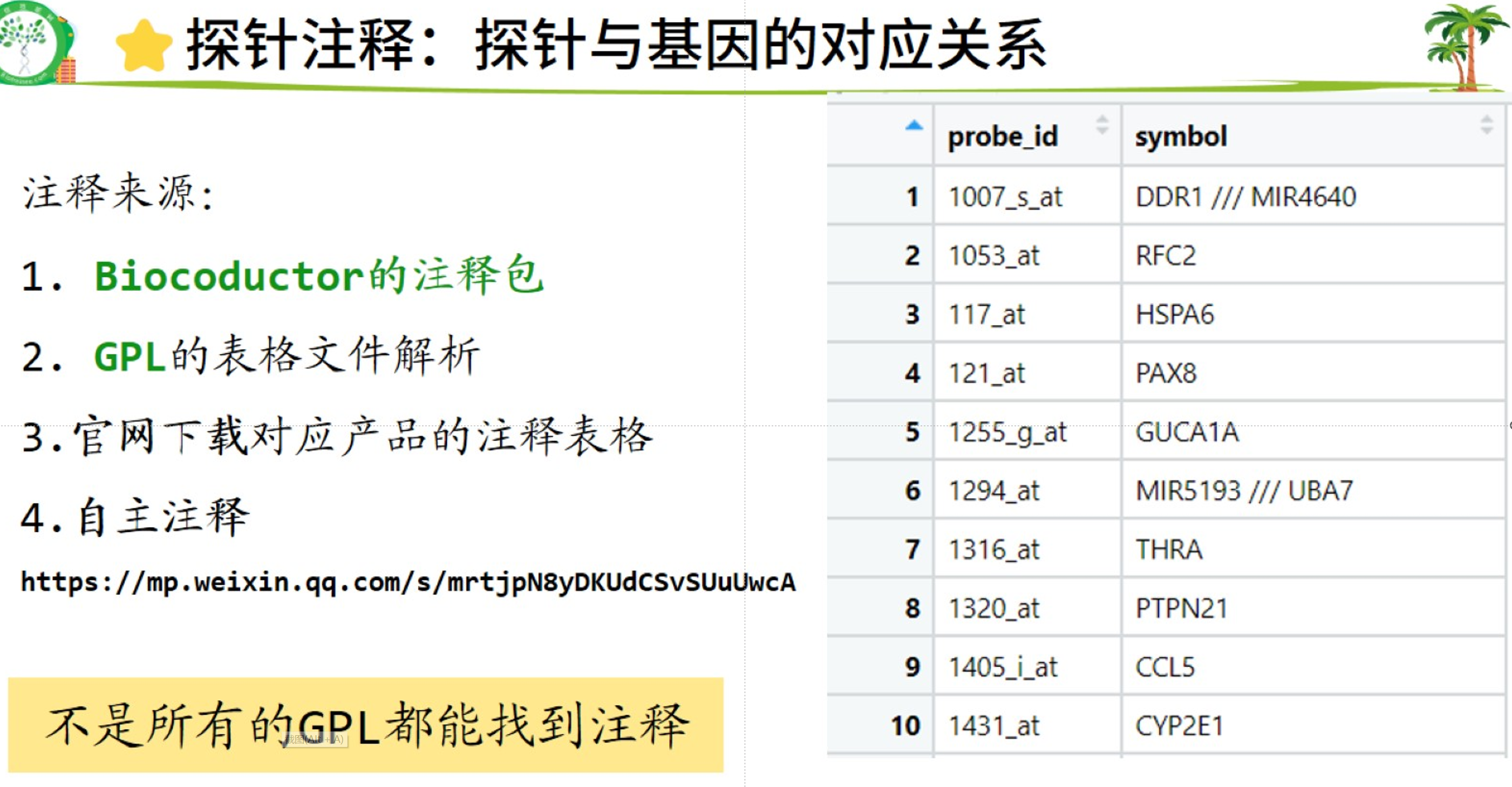
#2.探针注释的获取-----------------
#捷径,以下代码可以检索使用那种方法获得基因注释
library(tinyarray)
find_anno(gpl_number) #打出找注释的代码
ids <- AnnoProbe::idmap('GPL570')
表示两种代码都可以获取注释文件
#四种方法,方法1里找不到就从方法2找,以此类推。
#方法1 BioconductorR包(最常用)
gpl_number
#http://www.bio-info-trainee.com/1399.html
#获取了GPL编号后,登陆网站,搜索使用那个R包进行注释,注意R包名称后面有.db后缀
if(!require(hgu133plus2.db))BiocManager::install("hgu133plus2.db")
library(hgu133plus2.db)
ls("package:hgu133plus2.db") ##加载R包后,查看R包中哪部分是所需要的注释,R包无法自动补齐,注意
ids <- toTable(hgu133plus2SYMBOL) ##使用toTable函数加载R包中的SYMBOL,并生成数据框
head(ids)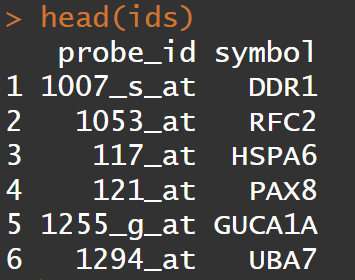
获取了一组探针和注释的数据框
# 方法2 读取GPL网页的表格文件,按列取子集
##https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
if(F){
#注:表格读取参数、文件列名不统一,活学活用,有的表格里没有symbol列,也有的GPL平台没有提供注释表格
#read.delim函数是read.table的替代函数,用来读取,sep默认\t,默认header=T
b = read.delim("GPL570-55999.txt",
check.names = F,
comment.char = "#")
colnames(b) ##返回列名用来复制列名
ids2 = b[,c("ID","Gene Symbol")]
colnames(ids2) = c("probe\_id","symbol")
k1 = ids2$symbol!="";table(k1) ##symbol列部分的空格为空字符串,取不要空格的行
k2 = !str_detect(ids2$symbol,"///");table(k2) ##
ids2 = ids2[ k1 & k2,]
# ids = ids2
}
##GPL网站下载的表格文件中可能存在多余的行,探针没有对应genesymbol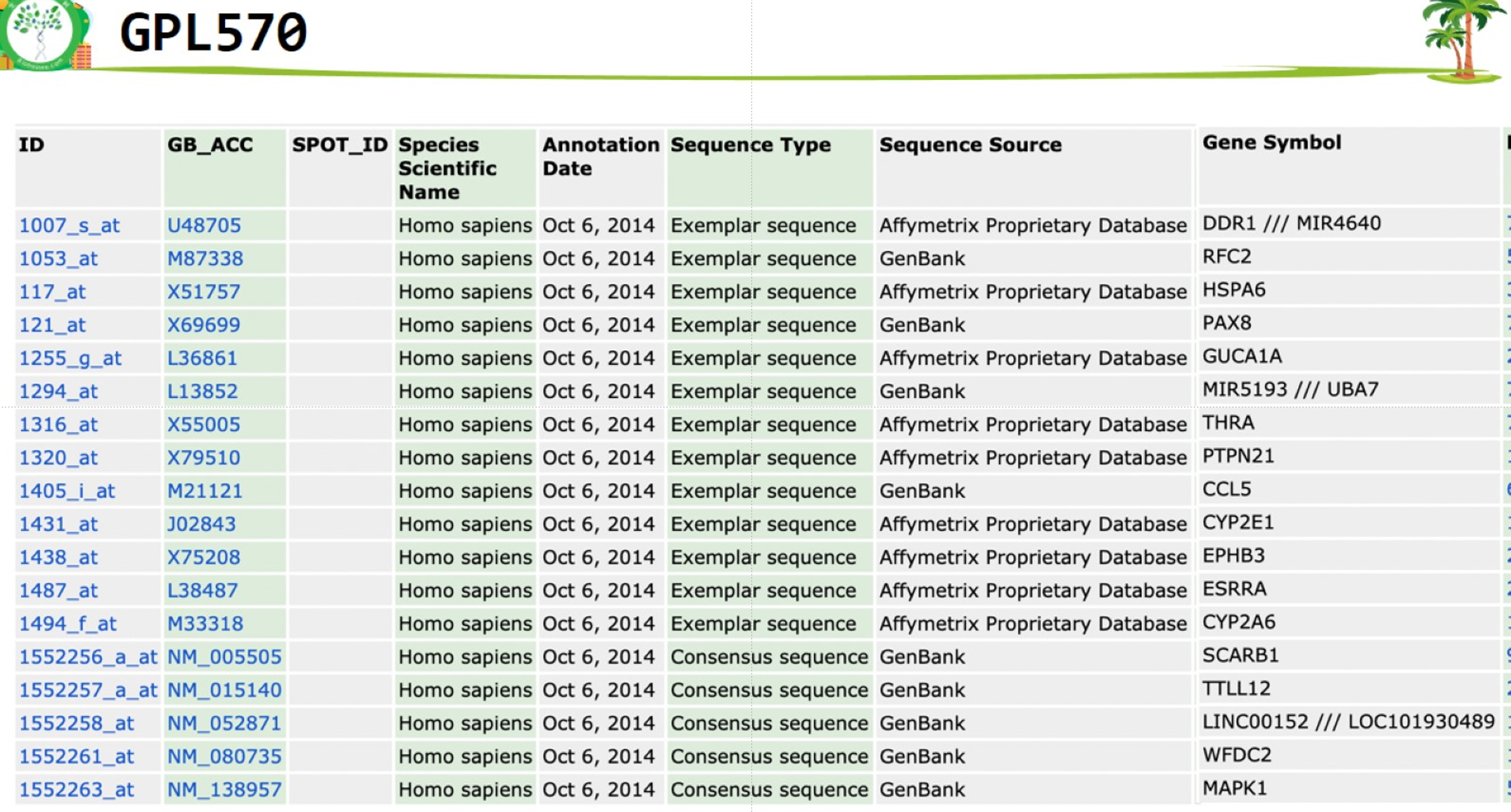
GPL570网页表格
理想情况下,表格中有gene symbol
有的表格中只有ensambleID等,需进一步转换成 gene symbol

GPL16956 网页表格
有些没有任何ID,只有探针序列,这种可以进行自主注释。
# 方法3 官网下载注释文件并读取
##http://www.affymetrix.com/support/technical/byproduct.affx?product=hg-u133-plus
# 方法4 自主注释
#https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
save(exp,Group,ids,gse_number,file = "step2output.Rdata")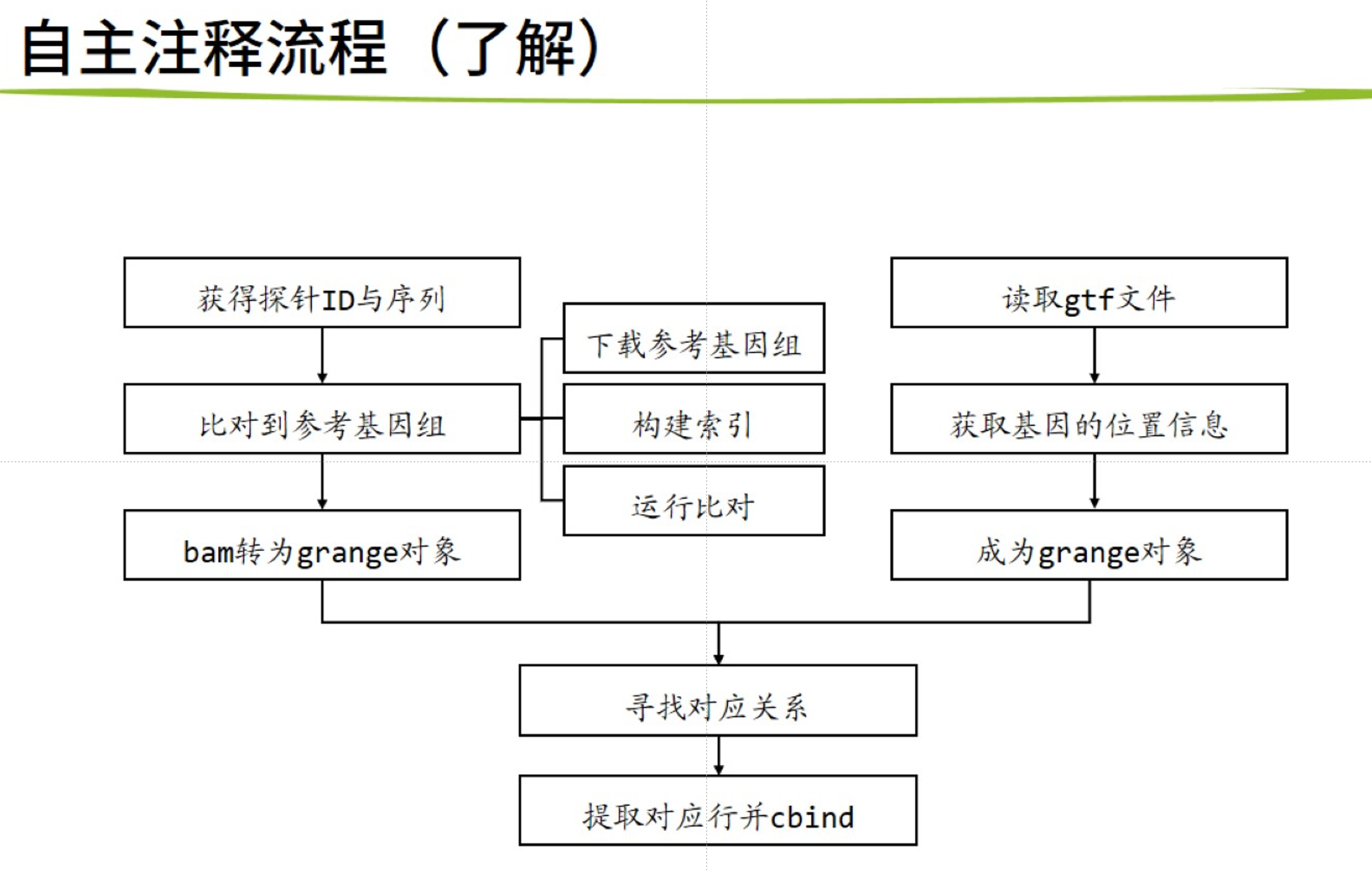
自主注释
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号