2023-11月的马拉松在线互动授课答疑精选

2023-11月的马拉松在线互动授课答疑精选

下面是优秀实习生的整理和分享
1【R包】有什么好办法可以把之前安的R包一次性全部清除,想全部重新安一遍。前几天安了一半报错没解决完,忘了安到哪了
有一些基础R包是不能清除的。想重新安装,把我们给学员准备工作的代码从第一行开始运行即可。
2【课程】回放啥时候可以看??
每天直播结束就会有回放,可以方便的快进快退复习我们的直播互动授课知识点,随时欢迎微信群图文并茂沟通哦!
3【R包】library(clusterProfiler)报错
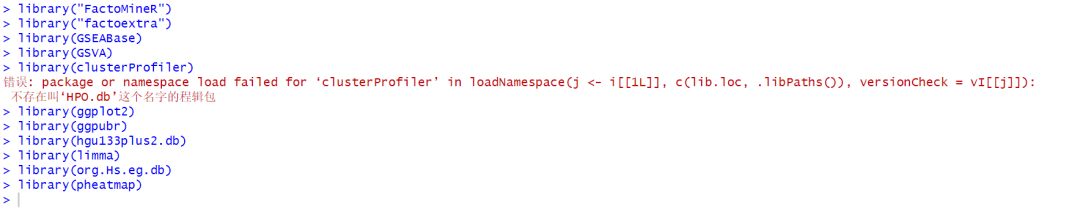
这个问题最近很常见了。安装新版本的 R 4.3 之后,安装 clusterProfiler 出现报错:依赖包不存在, 这其实是新版本的 bioconductor 3.18 在使用 clusterProfiler 的时候,引入了一个新的依赖包 HPO.db (还有 MPO.db),这个包在安装的时候,编译过程可能需要联网下载缓存数据。(但是,2024已经没有这个问题了)
该包出自 Y 叔实验室,在 github 上面有相关的 issue:https://github.com/YuLab-SMU/clusterProfiler/issues/606 亲测确实不好安装,本地服务器,开代理,服务器上都无法安装成功,怀疑是 R 包没有处理好细节,是 Y叔一个学生写的,使用本地安装会比较容易。
4【R包】安装的时候遇见这个error 该如何解决呢?
Error in askYesNo(msg) :
Unrecognized response “BiocManager::install(c("GEOquery","limma","impute" ),ask = F,update = F)”
你运行这句代码的时候,下面有提示,让你回答一个yes或者no,你没有回答。你一直在点run或者输入了别的。你重新运行回答一个yes就可以了。
5【R包】我重新下载了R后,hug133plus2这个包安装不上了
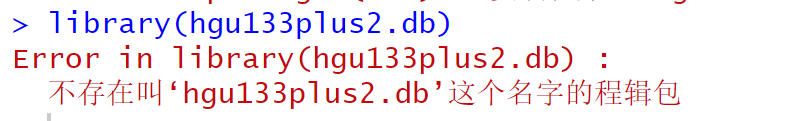
把这个包对应的安装代码重新运行一遍。
6【R包】HPO这个,我下载到本地了还是不行
你下载包括HPO和mpo一起的,你应该先解压出来,里面是两个gz文件,分别安装。
7【R包】请问这种情况需要做什么吗?
不用做,继续往下运行,最后library 没有关键词 error 即可。
8【R包】请问运行到最后是这样的,算成功了么?需不需要保存这份文件呀?
非常棒了,无需保存,静待授课。
9【R包】今天遇到的新问题是找不到RSQLite,我看了一下答疑,没找到相关的解答,报错如下
> library(GSEABase)
Loading required package: annotate
Loading required package: AnnotationDbi
Error: package or namespace load failed for ‘AnnotationDbi’ in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
there is no package called ‘RSQLite’
Error: package ‘AnnotationDbi’ could not be loaded
单独对你缺的这个包进行安装。
BiocManager::install(c("RSQLite" ),ask = F,update = F)
10【Rstudio】这样运行过的代码,绿色,蓝色,黑色的字是有不同的意思

好问题,是不同意思,第一天授课结束后你就会学会啦。
11【R安装】文档里说r和Rstudio需要安装在C盘,我之前安装在了其他盘了,我需要卸载重新安装吗?
如果你使用过一段时间,并且没有问题,就可以继续使用。但如果你安装完没有用过,建议卸载重装。
12【R包】请问这个报错是为什么?
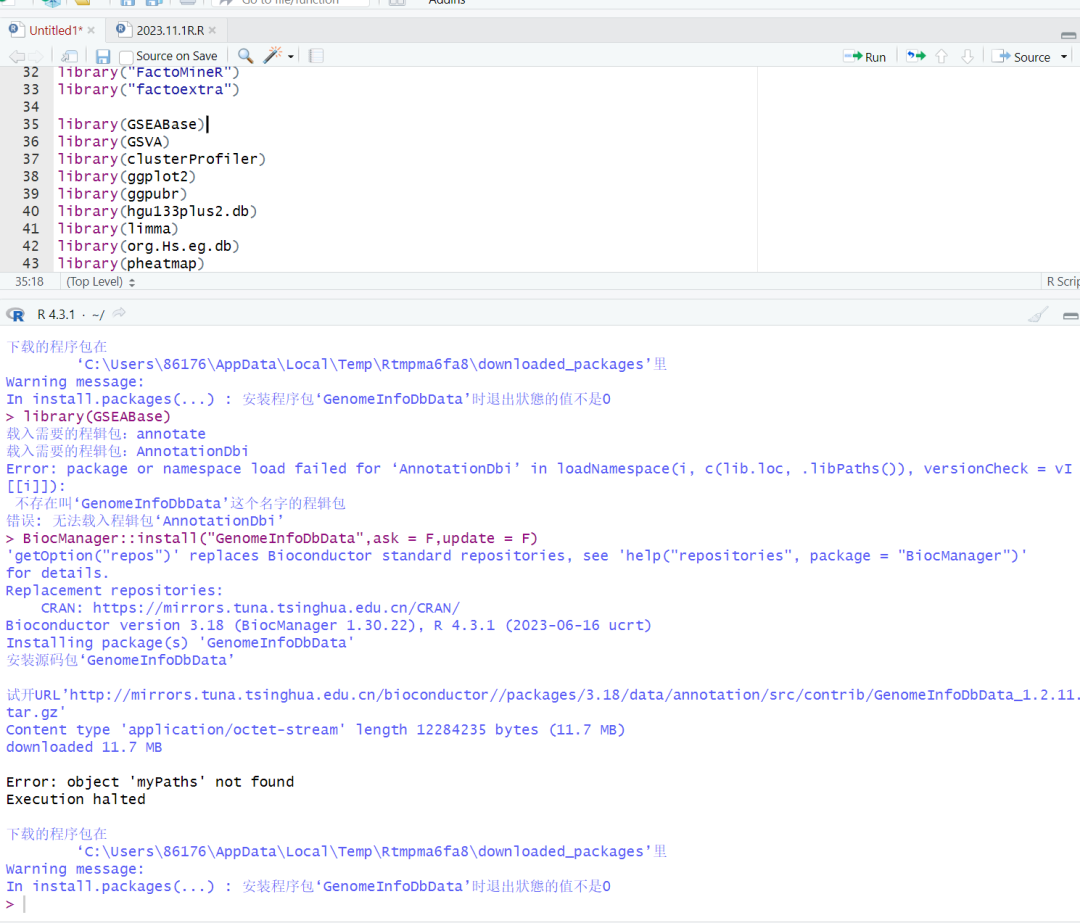
请依次检查
- 是否安装了Rtools;
- 有没有改过哪些系统设置?R语言和 Rstudio 有都安装到C盘吗?
13【R包】想问一下为什么分开了还是会提示这样?
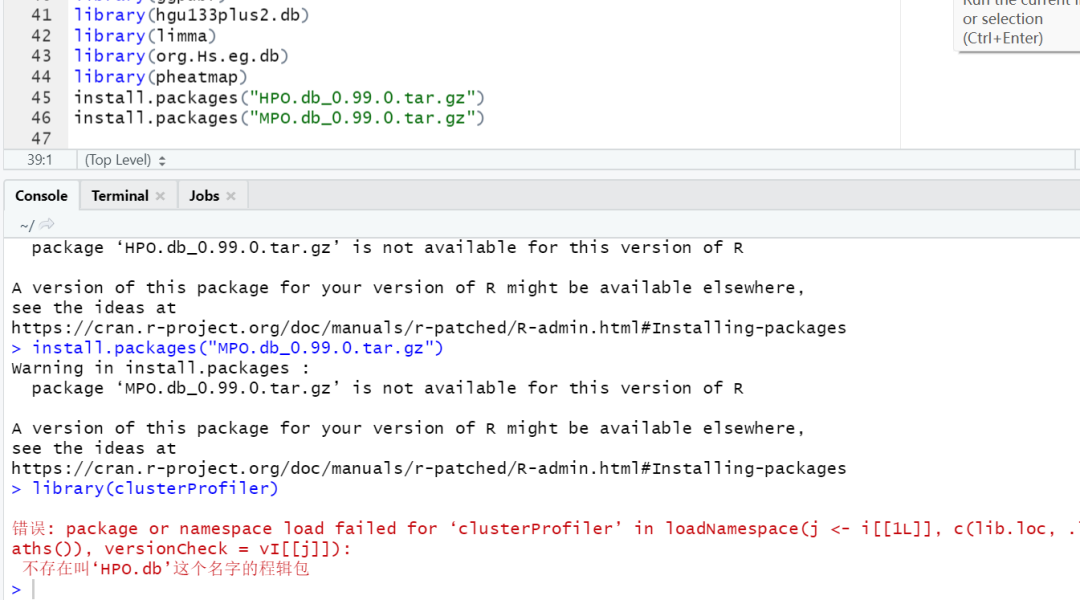
是路径问题,答疑文档最后一个,有提到如果你这两个 gz 下载之后放在桌面,在Rstudio里install 的时候,需要给对路径。
14【R包】为什么还是报错呀?
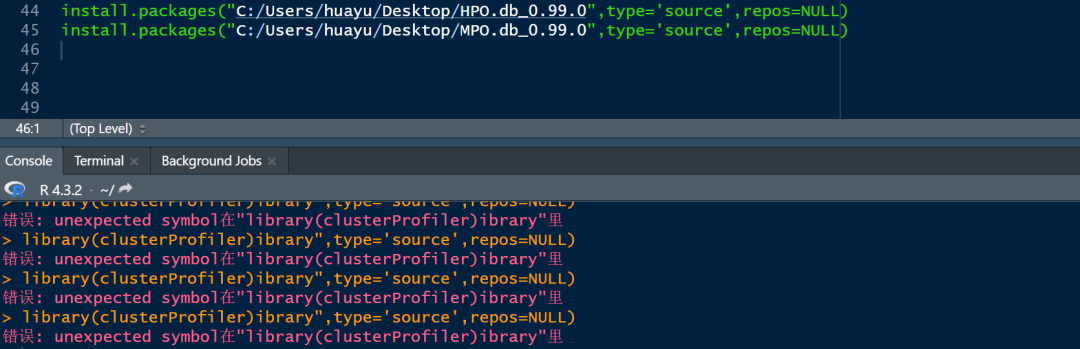
请检查一下是否安装了Rtools哦~
15【软件安装】这种情况怎么处理
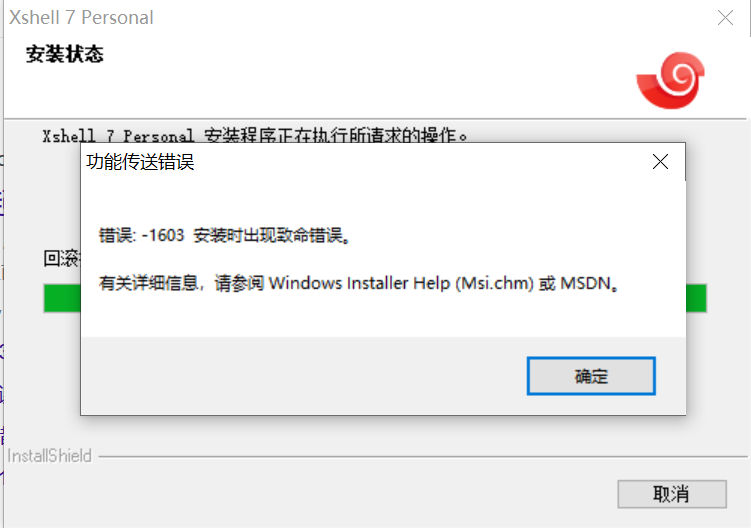
那安装Termius这个软件哈,课上也是主要用这个软件呢。
16【R包】这种可以吗?我的R是4.3.1的版本

没问题,不需要那么新。
17【R包】以前安装的4.2.2的R,够用了么?还是要重新安装
课后重新安装一下。
18【R包】请问一下这个是啥情况?
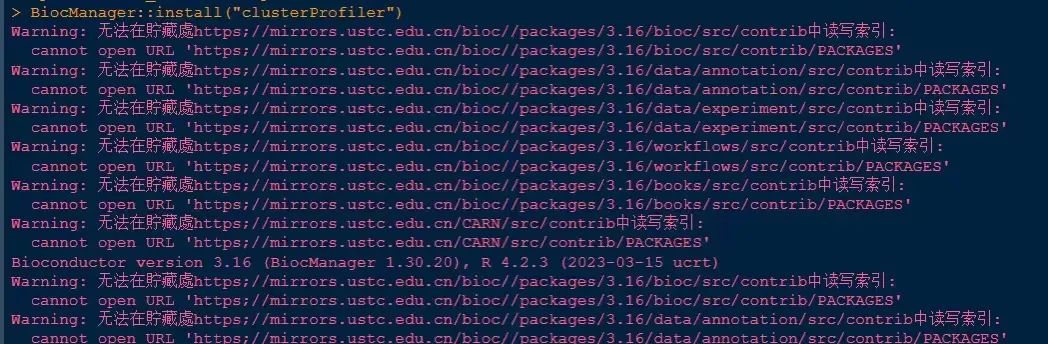
如果你对R语言版本没有要求,建议卸载重装新版本 R4.3。如果更新R版本,部分R包要重新安装,不过不难的。
19【R包】为什么我是b报错然后加引号后就能出结果,但a没加引号不报错?
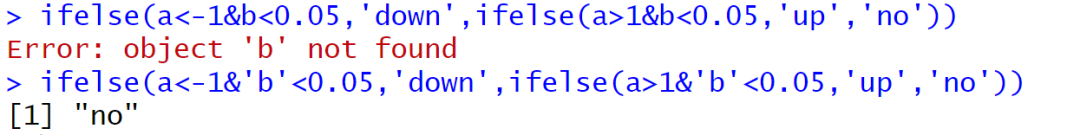
这里是因为a<-1 是程序识别为 a <- 1。<-是赋值符号。
20【R包】每次上课前都要library一遍么?
并不需要,按需library即可。
21【Rmarkdown】请问r markdown不能安装是什么原因?我是Mac电脑
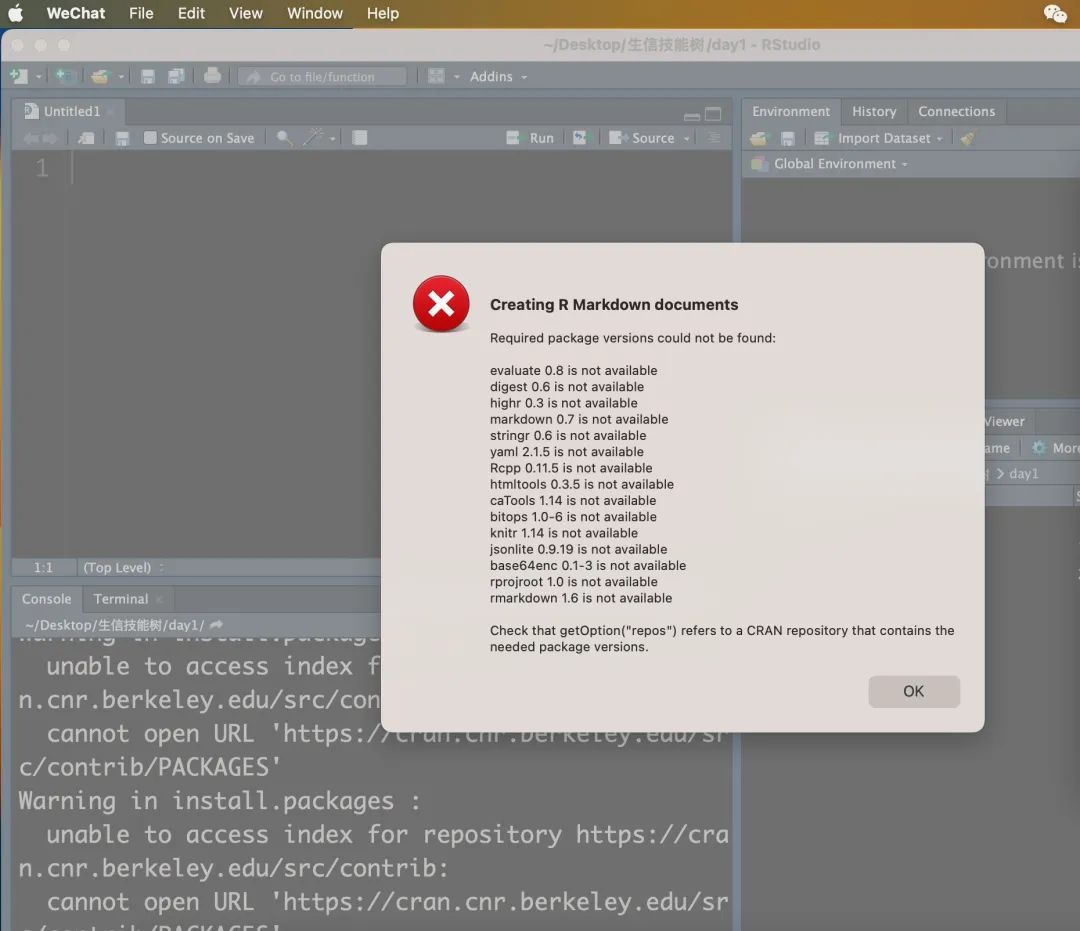
先运行这两句代码设置一下镜像
options("repos"= c(CRAN="https://mirrors.bfsu.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.bfsu.edu.cn/bioconductor/"),然后再install.packages("rmarkdown")
22【R包】install.packages和installed.packages区别在哪


23【R绘图】我拿R做列线图的时候发现体现不了中文,只有英文变量可以显示
中文无法识别。
24【R实战】为什么出现这个

拼写错误,P要大写!
25【R实战】这是为啥呢?
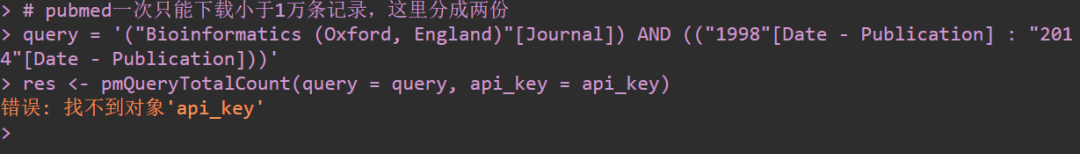
跑代码的时候是不是漏掉了赋值api_key呢?
26【R实战】是因为网络的问题报错吗?该怎么解决呢?
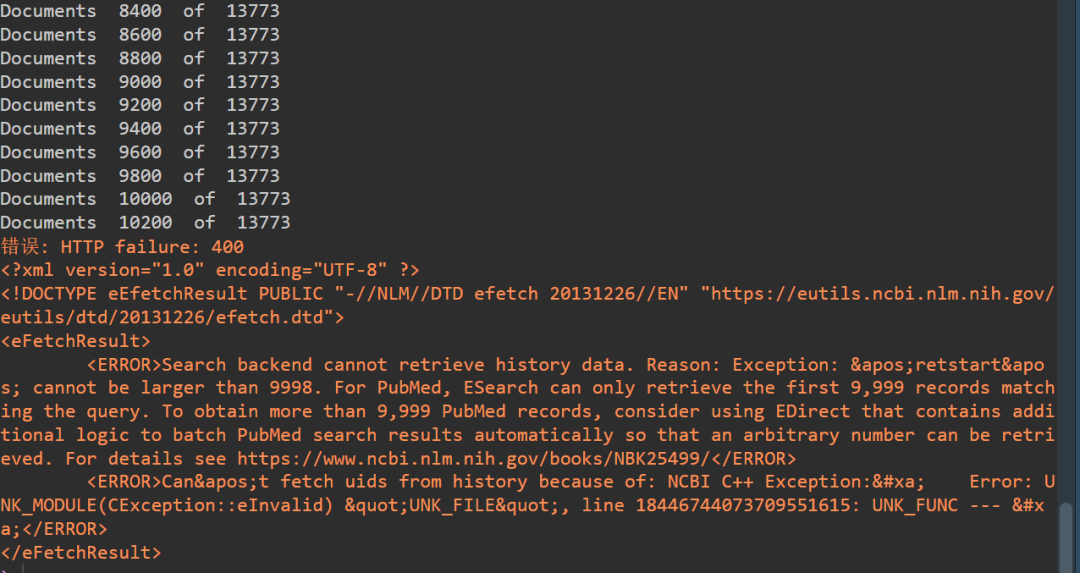
看起来是网络问题。pubmed一次只能下载小于1万条记录,尽量把抓取的数量调少一点。
27【R包】read.csv的说明文档里这三个点意思是剩余的内容同上吗?

是的。
28【Rstudio】请问内置数据的显示会出现不一致的情况吗?比如volcano这个矩阵。dim显示的是61列,没有问题,但是在数据窗口只能看到50列
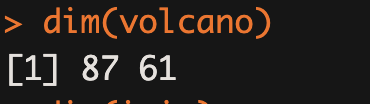
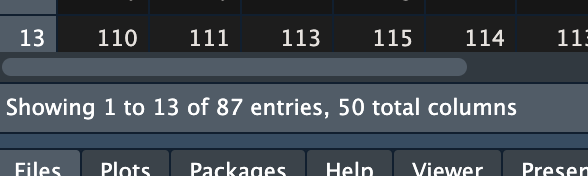
更新Rstudio即可,R语言可以不用更新。
29【Rstudio】在Rstudio的左上角输入代码后用什么快捷键run?我看小洁老师好像是回车就run了,我试了一下不行
Win的话 ctrl + 回车就可以代替run
Mac Command+回车
30【R包】请问这个怎么安装不成功啊
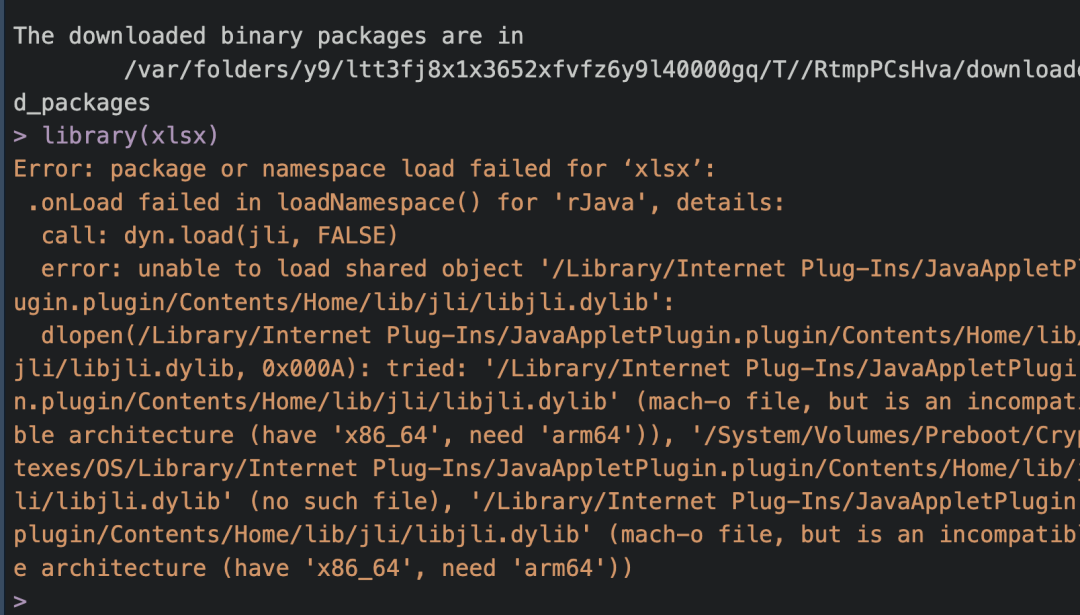
即便是你的Mac是M1,2,3芯片,安装R语言还是建议选用群公告云盘的x86版本。
31【R实战】请问这个报错是啥意思啊?我想要批量导出CSV文件,根据data文件其中一列的取值输出为每个文件的名字
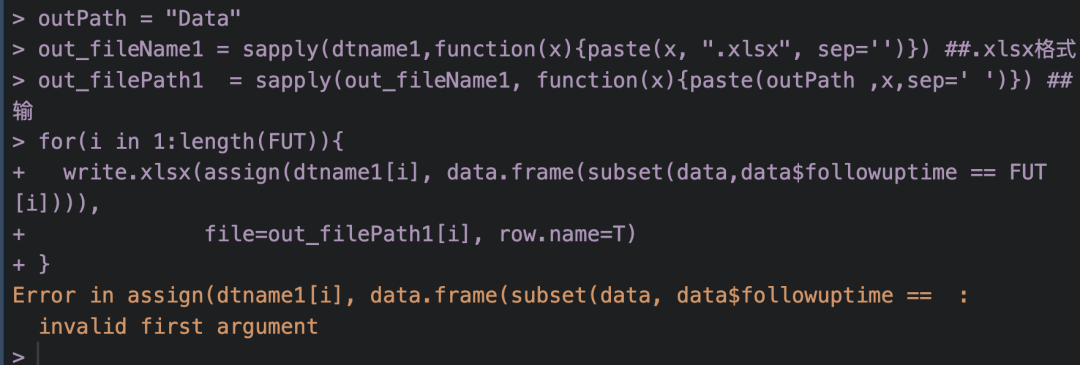
assign函数使用不对,建议查询一下参数,assign第一个参数得是字符。
32【R实战】不知道为啥这个GSE72713芯片数据集缺少表达矩阵的信息?
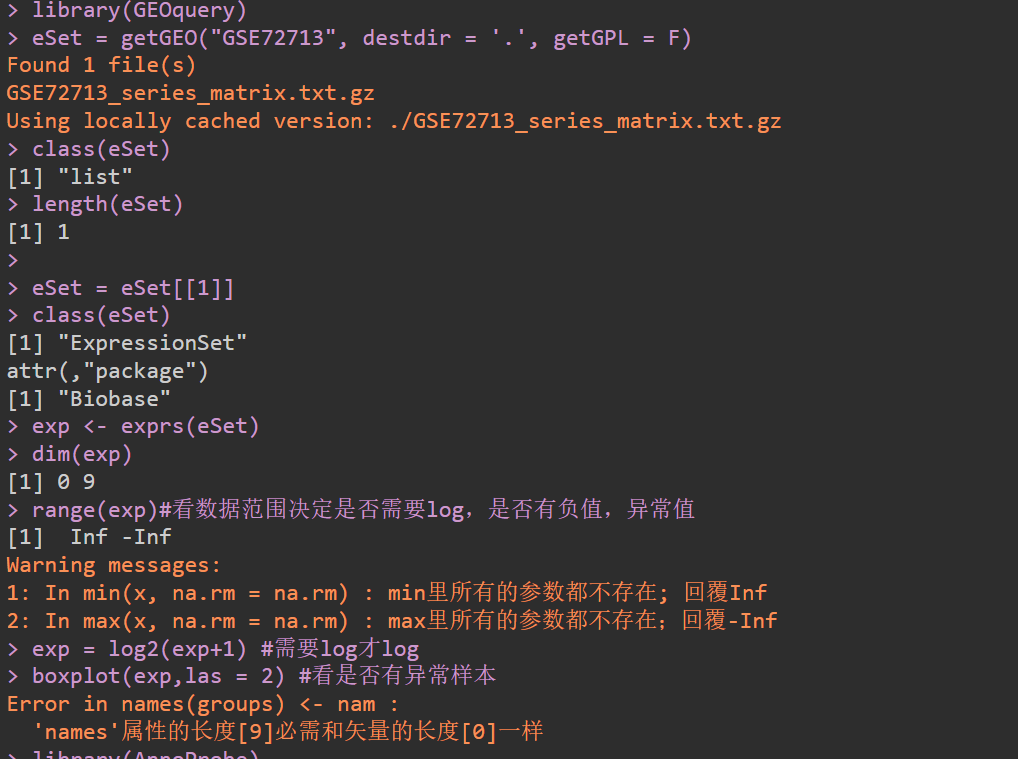
是转录组测序,你需要熟悉你的数据集。
33【R实战】这种数据是不是被加密了?有40多的呢
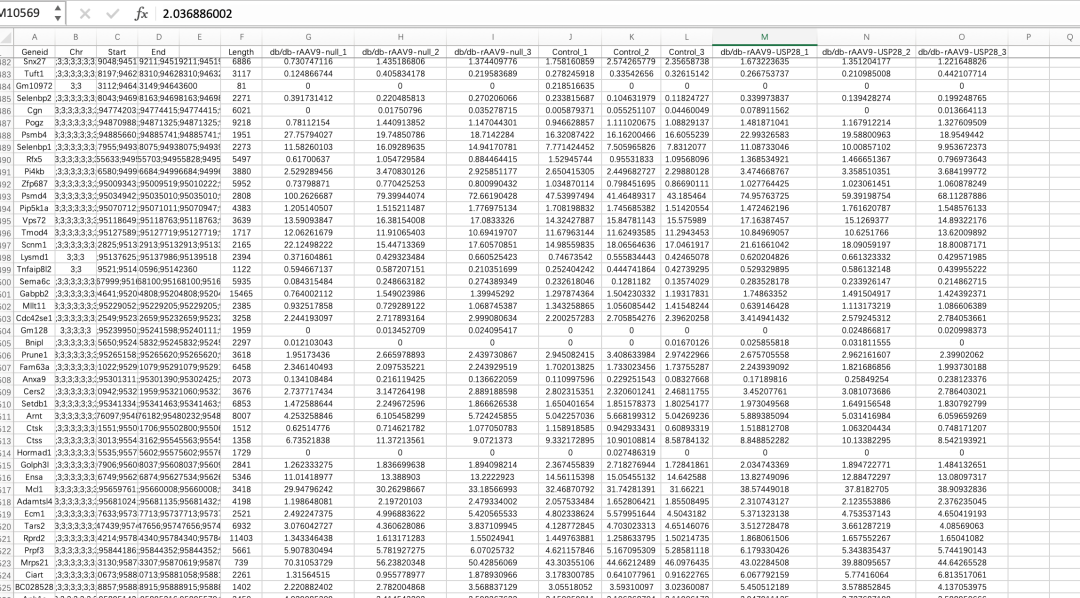
你的意思是认为40多就是加密,我可不是这么讲的啊,我说的加密指的是是标准化,有一半正数一半负数的。你这个不是。
34【软件安装】注册后显示14天使用期是正常的嘛……

是正常的,到时候你可以用学校的邮箱免费使用高级版本,或降级为低级版本,但只需要普通功能就可以完成全部的学习和正常使用了。
35【Linux】我在做01Linux基础的课后练习-场景五tar练习的第2题时,将“test.tar.gz"文件移动到”test"中并解压打开时会出现报错,前面的操作一切正常,请问一下这是什么原因呀?我在家目录下边写的命令,是不是要切换到test文件夹下再写命令
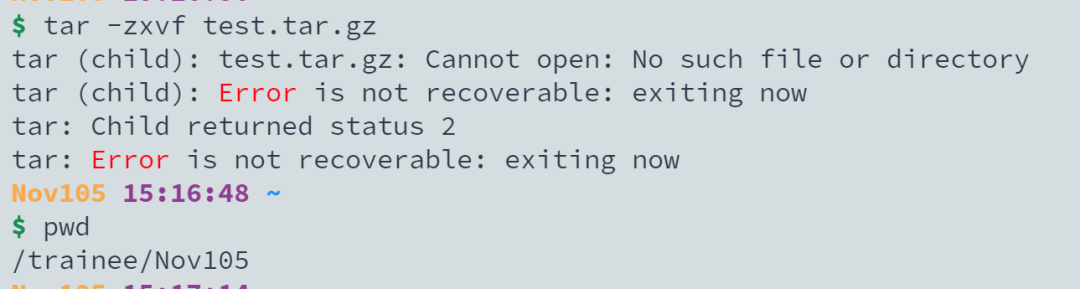
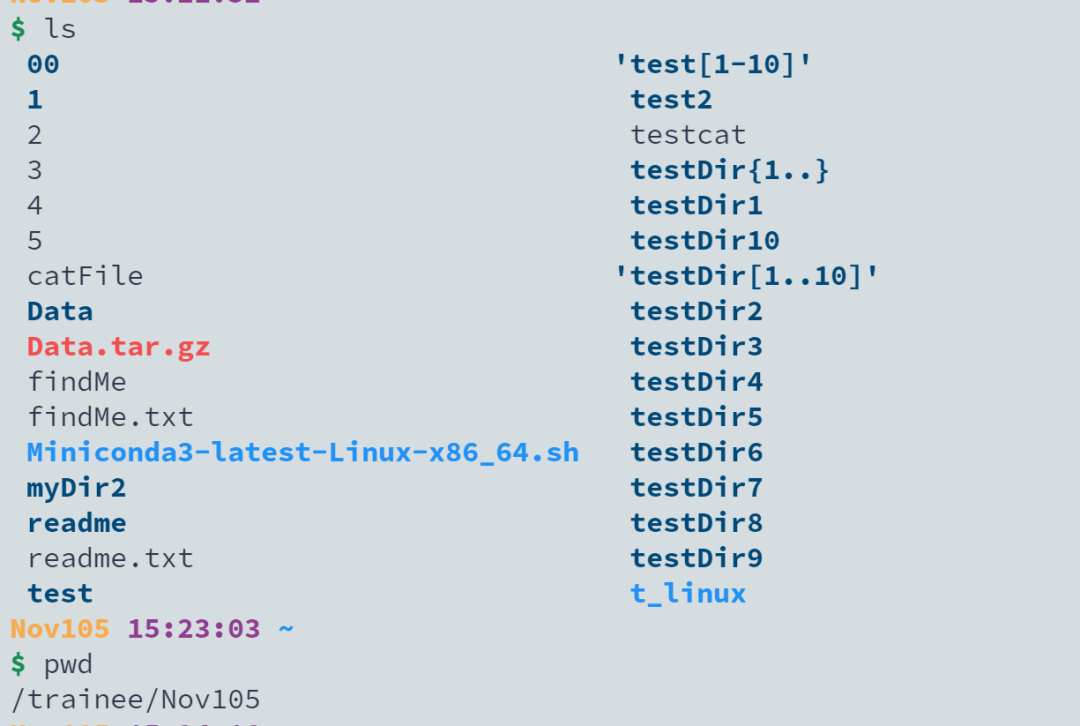
ls查看当前文件夹看一看,结合你ls 的结果和Cannot open: No such file or directory 你发现问题没有?
36【R实战】想请教一下gsea富集的话一般也是拿差异基因进行geneset的富集吗?我送公司测序分析回来的结果在gsea这部分他们是拿我所有测出来的基因进行富集的,这样合理吗……还有一个问题就是我用小洁老师gsea代码重新跑的差异基因gsea富集分析,但是就富集到一条通路,我看了GSEA函数的帮助文档,不确定这个显著通路到底是根据padj还是pvalue来进行筛选的呢?
是需要全部的基因做gsea哦,根据padj还是pvalue来进行筛选都不好,显著与否取决于你是否看重那个通路。如果你没有目标,就选择top即可。
37【R实战】KEGG富集报错
做KEGG的过程中,执行:kk <- enrichKEGG(gene=gene, organism="hsa", pvalueCutoff=1, qvalueCutoff=1) 报错:
我在脚本里添加了如下代码行:library(R.utils) R.utils::setOption("clusterProfiler.download.method","auto") 报错依旧,能帮忙看一下吗?谢谢!
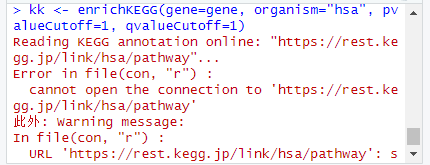
学员自查:网络的问题。其实也有可能是r包版本问题,有一个很简单的测试,就是如下所示代码:
lapply(c('clusterProfiler','enrichplot','patchwork'), function(x) {library(x, character.only = T)})
data(geneList, package="DOSE")
# Please go to https://yulab-smu.github.io/clusterProfiler-book/ for the full vignette.
kk2 <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 10000,
minGSSize = 10,
maxGSSize = 200,
pvalueCutoff = 0.05,
pAdjustMethod = "none" )
gseaplot2(kk2, geneSetID = rownames(kk2@result)[head(order(kk2@result$enrichmentScore))])
gseaplot2(kk2, geneSetID = rownames(kk2@result)[tail(order(kk2@result$enrichmentScore))])
这个是四句话代码,如果运行失败,说明大家的r包版本有问题哦,因为上面的代码是测试数据和示例代码,理论上任何人的任何的电脑设备都是联网就可以运行的。
38【软件安装】conda安装trim-galore时报错,安装命令为conda install trim-galore
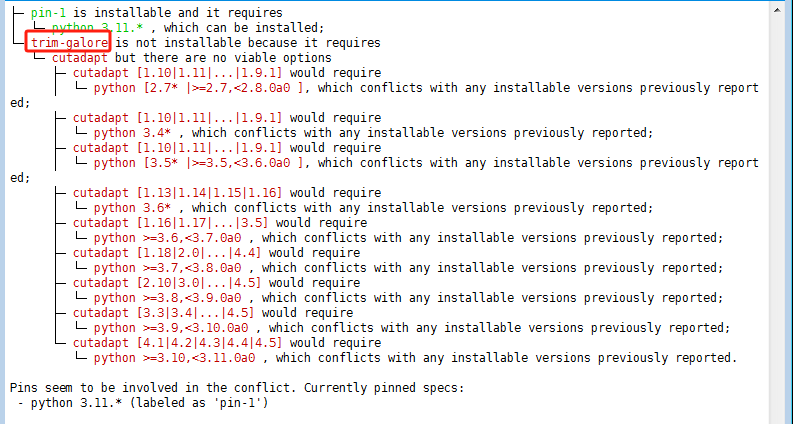
看这个报错是你需要先装上cutadapt,总的来讲,归根到底是python版本的问题。目测是cutadapt和base里的python不兼容 但是cutadapt没装上,进而导致 trim-galore的问题。python 3之间也有差别的。你创建个新的环境,用上课讲的办法直接导入试试。指定python版本为3.7或者3.8应该能避免问题。毕竟现在python都到3.12了,3.7已经是2018的事情了。
39【Linux实战】在统计多少条reads时,代码顺序为什么不是先除4并打印(awk '{print $0/4}'),然后再统计(wc -l)呢?


第一个代码是先用wc -l算出行数100000,在后面的awk里面被除以4的是行数100000,所以得出25000。
第二个代码中被除以4的是每一行的具体的值,除以4之后每一行只是计算出了新的值,总数还是100000。
40【Linux实战】这里的 ^M$ 和之前讲的 ^$ 区别在哪?
不同的编码方式,^M$ 是Windows下的。
41【转录组实战】关于Hisat2 比对数据,不生成中间文件直接生成bam文件的这句代码最后的占位符“-”有些疑问,我试着运行了一下有没有占位符的情况,并没有发现结果有什么不同欸

这个 - 是一个占位符,是告诉上一条命令,“上一步的输出要放到这个位置”,如果不设置的话默认是在最末尾的。之前版本的samtools如果不设置这个符号是有可能会报错的,因为samtools的有些命令的输入文件是在中间而不是末尾,因此要加上这个符号明确一下。
42【R实战】我有一组数据,横坐标是天数Days,但是非等距,我想要在R中作图,让它等距显示,并且把每个y对应的天数Days都在横坐标上显示出来,请问该如何实现呀?
可以将Days转化为character后合并到原数据框,factor转化这一个新的列。
43【R实战】这个报错是怎么回事啊
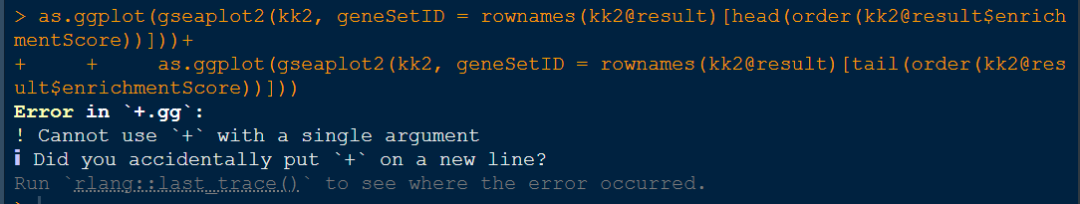
多加了一个加号。
腾讯云开发者
