全长转录组 | 三代全长转录组分析流程(PacBio & ONT )-- Flair
原创
全长转录组 | 三代全长转录组分析流程(PacBio & ONT )-- Flair
原创
三代测序说
发布于 2024-04-12 15:21:55
发布于 2024-04-12 15:21:55
今天我们介绍一款使用三代全长转录本数据进行转录本校正,聚类,可变剪切分析,定量和差异分析为一体的工具 - FLAIR。来自加利福尼亚大学圣克鲁斯分校(University of California,Santa Cruz)的Angela Brooks团队(图1)开发的全长可变转录本(isoform)分析工具FLAIR (Full-Length Alternative Isoform analysis of RNA),于2020年03月18号发表在《Nature Communications》杂志上,题目为 Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns。该工具可用来鉴定高可信度转录本,差异剪切事件分析和差异转录本异构体(isoform)分析。

图1. Angela Brooks团队
成熟mRNA前体(Pre-mRNA) 的剪接是由一种被称为剪接体(Spliceosome)的RNA-蛋白质复合物执行的。剪接体由 5 个小的核糖核蛋白颗粒(snRNPs,包括 U1、U2、U4、U5 和 U6)和非 snRNP因子组装而成。在这 5 个 snRNP中,U2 snRNP在内含子的识别和前体折叠的组装过程中起着重要作用。SF3B1是人体U2 snRNP的核心成分。在各种癌症中,剪接因子SF3B1中的突变已与基因剪接的特征性改变相关联。特别是,SF3B1中的复发性体细胞突变(在同一类疾病不同患者中反复出现的体细胞突变,recurrent somatic mutations)已与多种疾病相关联,包括慢性淋巴细胞白血病(Chronic Lymphocytic Leukemia,CLL)、葡萄膜黑色素瘤(Uveal Melanoma)、乳腺癌( Breast Cancer)和骨髓增生异常综合征( Myelodysplastic Syndromes)。虽然已知SF3B1基因中的体细胞突变会导致基因剪接发生变化,但识别全长转录本异构体(isoform)的变化可能会更好地阐明这些突变的功能后果。
本文选取3个无SF3B1突变CLL患者样本(CLL - SF3B1WT)、3个SF3B1K700E 突变CLL患者样本(CLL - SF3B1K700E) 和3个普通B淋巴细胞样本为研究对象,通过三代测序 Oxford Nanopore(ONT)技术平台进行全长转录组测序,并为此开发了FLAIR分析流程用于识别高可信度转录本,进行差异剪接事件和差异转录本异构体(isoform)分析。利用三代数据,作者证实了SF3B1突变与差异性3'剪接位点的变化相关,与先前的研究结果一致。还观察到与SF3B1突变相关的内含子保留事件的明显下调。全长转录本分析将多个可变剪接事件联系在一起,可以更好地估计有效与无效异构体(isoform)的丰度。此项工作展示了纳米孔测序在癌症和转录本剪接研究中的潜在实用性(图2)。

图2. FLAIR文章摘要
一、软件介绍
FLAIR除了单独使用三代测序数据,也支持二代短读长测序数据,用以辅助增加识别剪切位点的准确度。FLAIR通过多步比对和剪切位点过滤以增加isoform识别的可信度,降低数据质量引起信号噪音的影响。FLAIR通过算法设计能够从三代ONT数据中识别微小的剪切变化。FLAIR软件一共六个大模块(modules),flair align,flair correct,flair collapse,flair quantify,flair diffExp和flair diiffSplice (图3)。
flair align:将三代测序序列与参考基因组进行比对。flair correct:根据参考基因组注释文件对剪切位点进行校正。如果提供二代测序数据,可进一步进行纠错校正。flair collapse: 将校正后的序列进行聚类和合并,最终形成来源于样本的高可信度转录本参考序列。对于所有实验重复/不同条件处理的样本经过上一步序列校正后,在这一步进行整合聚类和合并。flair quantify:对所有样本中的isoforms进行表达定量,生成表达矩阵。flair diffExp:在指定分组的情况下,对组间进行差异表达分析。flair diiffSplice:对组间差异可变剪切事件进行分析。
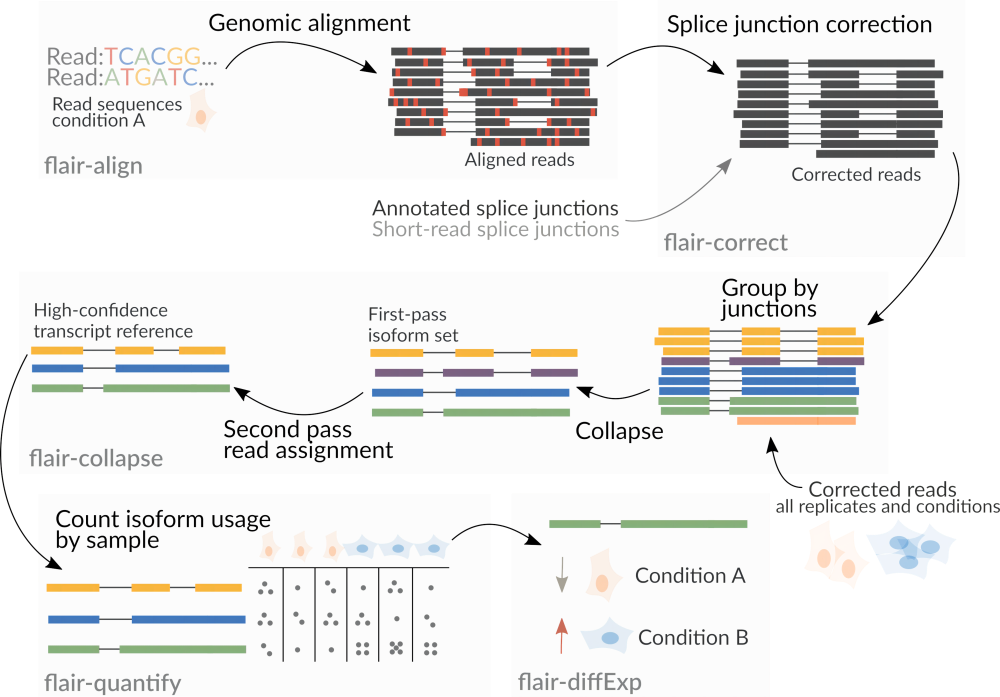
图3. FLAIR软件分析流程
建议在运行flair collapse步骤之前合并所有样本的校正序列(psl 或 bed文件 ),这样方便于后面的定量。bed12 和 psl 文件之间可以利用 kentUtils 中的bedToPsl 或 pslToBed命令进行转换。
二、软件安装
Flair v2.0 - 2023.6.14
官方github网址:https://github.com/BrooksLabUCSC/flair
操作文档:https://flair.readthedocs.io/en/latest/
- 最方便和简单的方法还是利用
conda进行安装。
#创建Flair软件环境,并安装
$ conda create -n flair -c conda-forge -c bioconda flair
$ conda activate flair
$ flair [align/correct/...]- 支持
docker镜像。
$ docker pull brookslab/flair:latest
$ docker run -w /usr/data -v [your_path_to_data]:/usr/data brookslab/flair:latest flair [align/correct/...]三、软件使用
上面我们提到FLAIR包含多个模块,需要依次运行
1. flair align
输入文件为:
- 参考基因组:
ref.fa。 - 三代测序数据:
reads.fq或reads.fa。
$ flair align -g genome.fa -r <reads.fq>|<reads.fa> [options]此模块使用minimap2软件对三代测序数据与提供的参考基因组(ref.fa)进行比对,最后会将SAM文件转化为BED12文件,同时会保留比对BAM文件。
输出文件:
flair.aligned.bamflair.aligned.bam.baiflair.aligned.bed
选项:
- 必要选项
--reads Raw reads in fasta or fastq format. This argument accepts multiple
(comma/space separated) files. # 下机测序序列,接受.fasta 或 .fastq格式文件;多个文件可以逗号/空格分开。
At least one of the following arguments is required (至少提供一个):
--genome Reference genome in fasta format. Flair will minimap index this file
unless there already is a .mmi file in the same location. # 基因组参考序列(.fa),minimap会自动建立索引。
--mm_index If there already is a .mmi index for the genome it can be supplied
directly using this option. # 可以直接输入minimap索引文件 .mmi。- 可选选项
--help Show all options. #帮助命令。
--output Name base for output files (default: flair.aligned). You can supply
an output directory (e.g. output/flair_aligned) but it has to exist;
Flair will not create it. If you run the same command twice, Flair
will overwrite the files without warning. #输出文件夹路径和文件前缀,文件夹必须自己创建。
--threads Number of processors to use (default 4). #线程,默认为4。
--junction_bed Annotated isoforms/junctions bed file for splice site-guided
minimap2 genomic alignment. #异构体/连接位点注释的bed文件,用于辅助minimap基因组比对。
--nvrna Use native-RNA specific alignment parameters for minimap2 (-u f -k 14) #RNA直接测序选项。
--quality Minimum MAPQ score of read alignment to the genome. The default is 1,
which is the lowest possible score. #比对的MAPQ值,默认为最低的1。
-N Retain at most INT secondary alignments from minimap2 (default 0). Please
proceed with caution, changing this setting is only useful if you know
there are closely related homologs elsewhere in the genome. It will
likely decrease the quality of Flair's final results. #保留几个次好的比对结果,默认为0。
--quiet Dont print progress statements. #不输出过程。注释:
- 如果对人类全长转录本进行分析,建议最好使用Heng Li推荐的人类参考基因组 GCA_000001405.15_GRCh38_no_alt_analysis_set,详细参见Heng Li的博客。
- 如果下机数据来自 Oxford Nanopre(ONT)平台,建议对原始数据使用Pychopper后(目的是鉴定全长转录组本),再运行FLAIR。
- 如果测序数据已经比对过了,可以使用
bam2Bed12将bam文件转换为bed12,然后再运行flair correct。 - 关于
--nvrna选项设置,可以参考minimap2文档。 - 关于比对质量MAPQ: MAPQ scores。
2. flair correct
输入文件:
- 上一步比对后的bed文件:
query.bed12。 - 参考基因组:
ref.fa。 - 基因组注释文件:
ref.gtf。 - 内含子界定文件(可选):
introns.tab。
usage: flair correct -q query.bed12 [-f annotation.gtf]|[-j introns.tab] -g genome.fa [options]输出文件:
<prefix>_all_corrected.bed校正序列的bed文件,供下一模块使用。<prefix>_all_inconsistent.bed舍弃的比对序列。<prefix>_cannot_verify.bed如果所在染色体没有注释,序列会放入此文件。
选项:
- 必要选项
--query Uncorrected bed12 file, e.g. output of flair align. #上一步比对后的bed12文件。
--genome Reference genome in fasta format. #基因组参考文件。
At least one of the following arguments is required:
--shortread Bed format splice junctions from short-read sequencing. You can
generate these from SAM format files using the junctions_from_sam
program that comes with Flair. # 来自于二代短读长测序的剪切位点(bed文件格式),可以利用FLAIR自带的junctions_from_sam脚本,将比对后生成的SAM文件转化为BED文件。
--gtf GTF annotation file. #基因组注释文件。- 可选选项
--help Show all options #帮助命令。
--output Name base for output files (default: flair). You can supply an
output directory (e.g. output/flair) but it has to exist; Flair
will not create it. If you run the same command twice, Flair will
overwrite the files without warning. #输出文件夹路径和文件前缀,文件夹必须自己创建。
--threads Number of processors to use (default 4). #线程,默认为4。
--nvrna Specify this flag to make the strand of a read consistent with
the input annotation during correction. #RNA直接测序选项。
--ss_window Window size for correcting splice sites (default 15). #校正剪切位点的‘窗口(范围)’大小,默认值为15。
--print_check Print err.txt with step checking. #输出报错信息。3. flair collapse
输入文件:
- 上一步经过校正的转录本bed文件:
<prefix>_all_corrected.bed。 - 参考基因组:
ref.fa。 - 第一步提供的三代测序数据:
reads.fq或reads.fa。。
usage: flair collapse -g genome.fa -q <query.bed> -r <reads.fq>/<reads.fa> [options]- 通过校正后的序列定义的高可信度isoforms。因为FLAIR没有利用注释文件去合并isoforms,FLAIR将会以和isoform具有共有剪切位点链的名字命名序列。建议提供使用
--gft选项提供注释文件,这样FLAIR识别的isoforms可以以注释文件中相匹配的isoforms的名字进行重命名(gtf文件中transcript_id里的名称)。 - 这一步产生的中间文件会被删除,如果想保留则可以使用
--keep_intermediate,并且使用--temp_dir提供存储路径。 - 如果有多个样本,经过校正的序列
bed文件需要整合,然后才能运行flair-collapse。另外,所有原始的fasta或fastq文件用--reads指定,样本之间用空格/逗号分开,或者合并成一个文件。 - 请注意,
flair collapse暂时还不能处理较大的bed文件 (>1G)。如果发现FLAIR占用太多内存,可以将bed文件按照染色体分开,然后分别运行。
输出文件:
isoforms.bedisoforms.gtfisoforms.fa
选项:
- 必要选项
--query Bed file of aligned/corrected reads #比对完成/经过校正的序列
--genome FastA of reference genome #参考基因组
--reads FastA/FastQ files of raw reads, can specify multiple files #原始三代测序数据fasta/fastq, 可以指定多个。- 可选选项
--help Show all options. #帮助
--output Name base for output files (default: flair.collapse). #命名输出文件,默认为flair.collapse。
You can supply an output directory (e.g. output/flair_collapse) #指定输出文件夹。
--threads Number of processors to use (default: 4). #线程数,默认为4。
--gtf GTF annotation file, used for renaming FLAIR isoforms to
annotated isoforms and adjusting TSS/TESs. #gtf注释文件,用于FLAIR聚类的isoform的重新命名,调整转录本起始和结束位点。
--generate_map Specify this argument to generate a txt file of read-isoform
assignments (default: not specified).#生成序列对应isoform的文本文件,默认不指定。
--annotation_reliant Specify transcript fasta that corresponds to transcripts
in the gtf to run annotation-reliant flair collapse; to ask flair
to make transcript sequences given the gtf and genome fa, use
--annotation_reliant generate. #产生相应的转录本fasta序列文件。- 支持序列参数选项
--support Minimum number of supporting reads for an isoform; if s < 1,
it will be treated as a percentage of expression of the gene
(default: 3). #最小几个序列支持一个isoform,默认为3。
--stringent Specify if all supporting reads need to be full-length (80%
coverage and spanning 25 bp of the first and last exons). #支持序列必须都是全长(80%的覆盖率,第一个和最后一个外显子至少有25个碱基)
--check_splice Enforce coverage of 4 out of 6 bp around each splice site and
no insertions greater than 3 bp at the splice site. Please note:
If you want to use --annotation_reliant as well, set it to
generate instead of providing an input transcripts fasta file,
otherwise flair may fail to match the transcript IDs.
Alternatively you can create a correctly formatted transcript
fasta file using gtf_to_psl #至少覆盖剪切位点6个中的4个,插入序列不能大约3bp。
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation. #如果序列来自长序列建库方法(最小程度的打断)
--quality Minimum MAPQ of read assignment to an isoform (default: 1). #序列归为isoform最低的MAPQ值。- 变异选项
--longshot_bam BAM file from Longshot containing haplotype information for each read. #包含单倍型信息的BAM文件。
--longshot_vcf VCF file from Longshot. #包含变异信息的VCF文件。关于Longshot variant caller,请参考github page。
- 转录本起始和终止
--end_window Window size for comparing transcripts starts (TSS) and ends
(TES) (default: 100). #比较转录本起始和终止的窗口大小,默认为100。
--promoters Promoter regions bed file to identify full-length reads. #启动子区域的bed文件以鉴定全长序列。
--3prime_regions TES regions bed file to identify full-length reads. #转录本终止区域的bed文件以鉴定全长序列。
--no_redundant <none,longest,best_only> (default: none). For each unique
splice junction chain, report options include:
- none best TSSs/TESs chosen for each unique
set of splice junctions #对每一个剪切位点选择最好的起始和终止。
- longest single TSS/TES chosen to maximize length #选择最长的。
- best_only single most supported TSS/TES #单个支持最多的。
--isoformtss When specified, TSS/TES for each isoform will be determined
from supporting reads for individual isoforms (default: not
specified, determined at the gene level). #每一个isoform的转录起始和终止由支持它的序列所决定。
--no_gtf_end_adjustment Do not use TSS/TES from the input gtf to adjust
isoform TSSs/TESs. Instead, each isoform will be determined
from supporting reads. #不使用注释文件去校正isoform的起始和终止。
--max_ends Maximum number of TSS/TES picked per isoform (default: 2). #每一个isoform选中的TSS/TES的最大数值,默认为2。
--filter Report options include:
- nosubset any isoforms that are a proper set of
another isoform are removed #同时归为其它类的isoform被去除。
- default subset isoforms are removed based on support #基于支持数值去除isoforms子集。
- comprehensive default set + all subset isoforms
- ginormous comprehensive set + single exon subset
isoforms- 其它选项
--temp_dir Directory for temporary files. use "./" to indicate current
directory (default: python tempfile directory). #指定临时文件夹。
--keep_intermediate Specify if intermediate and temporary files are to
be kept for debugging. Intermediate files include:
promoter-supported reads file, read assignments to
firstpass isoforms. #保留中间文件。
--fusion_dist Minimium distance between separate read alignments on the
same chromosome to be considered a fusion, otherwise no reads
will be assumed to be fusions. #融合基因在同一个染色体上的分开距离。
--mm2_args Additional minimap2 arguments when aligning reads first-pass
transcripts; separate args by commas, e.g. --mm2_args=-I8g,--MD.
--quiet Suppress progress statements from being printed. #过程不输出。
--annotated_bed BED file of annotated isoforms, required by --annotation_reliant.
If this file is not provided, flair collapse will generate the
bedfile from the gtf. Eventually this argument will be removed. #提供isoforms注释的BED文件。
--range Interval for which to collapse isoforms, formatted
chromosome:coord1-coord2 or tab-delimited; if a range is specified,
then the --reads argument must be a BAM file and --query must be
a sorted, bgzip-ed bed file. #isoforms合并间隔。建议使用命令:
人
$ flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --check_splice --generate_map --annotation_reliant generate酵母
$ flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --no_gtf_end_adjustment --check_splice --generate_map --trust_ends4. flair quantify
输入文件:
- 样本,分组和数据路径:
reads_manifest.tsv。 - 来自上一步的isoform序列文件:
isoforms.fa。
usage: flair quantify -r reads_manifest.tsv -i isoforms.fa [options]输出文件:
样本isoform表达矩阵,可以用于后续flair_diffExp 和 flair_diffSplice。
选项:
- 必要选项
--isoforms Fasta of Flair collapsed isoforms #最终合并的isoform序列文件,来自flair collapse。
--reads_manifest Tab delimited file containing sample id, condition, batch,
reads.fq, where reads.fq is the path to the sample fastq file. #制表符分隔的样本id,实验条件分组,实验批次,测序数据(reads.fq)的路径。reads_manifest.tsv格式示例:
sample1 condition1 batch1 mydata/sample1.fq
sample2 condition1 batch1 mydata/sample2.fq
sample3 condition1 batch1 mydata/sample3.fq
sample4 condition2 batch1 mydata/sample4.fq
sample5 condition2 batch1 mydata/sample5.fq
sample6 condition2 batch1 mydata/sample6.fq注意:前三列命名的时候不要出现下划线。
- 可选选项
-help Show all options #帮助命令
--output Name base for output files (default: flair.quantify). You
can supply an output directory (e.g. output/flair_quantify). #指定输出文件前缀和路径。
--threads Number of processors to use (default 4). #线程,默认为4。
--temp_dir Directory to put temporary files. use ./ to indicate current
directory (default: python tempfile directory). #临时文件存放路径。
--sample_id_only Only use sample id in output header instead of a concatenation
of id, condition, and batch. #在表达矩阵表头只显示样本名称,而不是id,分组,批次都显示。
--quality Minimum MAPQ of read assignment to an isoform (default 1). #测序序列指定(归类)到isoform时的最小MAPQ,默认值为1。
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation. #如果序列来自长序列建库方法(最小程度的打断)。
--generate_map Create read-to-isoform assignment files for each sample. #生成序列对应isoform的文本文件,默认不指定。
--isoform_bed isoform .bed file, must be specified if --stringent or
--check-splice is specified. #isoform的bed文件。如果指定--stringent和--check-splice,这必须提供。
--stringent Supporting reads must cover 80% of their isoform and extend
at least 25 nt into the first and last exons. If those exons
are themselves shorter than 25 nt, the requirement becomes
'must start within 4 nt from the start' or 'end within 4 nt
from the end'. #支持序列必须都是全长(80%的覆盖率,第一个和最后一个外显子至少有25个碱基)。
--check_splice Enforces coverage of 4 out of 6 bp around each splice site
and no insertions greater than 3 bp at the splice site. #至少覆盖剪切位点6个中的4个,插入序列不能大约3bp。- 其它信息
后续flair_diffExp 和 flair_diffSplice需要表达矩阵样本表头信息包含id,分组和批次信息。所以建议一般不使用--sample_id。
5. flair diffExp
输入文件:
- 转录本定量表达矩阵:
counts_matrix.tsv。
usage: flair_diffExp -q counts_matrix.tsv --out_dir out_dir [options]这个模块对两个分组,每个分组可以有3个或者3个以上的重复实验数据进行差异'isoform表达'和'isoform使用'的分析。
- FLAIR使用 DESeq2 同时进行基因(gene)和转录本异构体(isoform)水平上的表达差异分析。
- FLAIR使用 DRIMSeq 只对转录本异构体(isoform)的差异使用(usage)进行分析。通过检测两个分组条件之间异构体(isoform) 的比例。
如果没有实验重复,可以使用diff_iso_usage进行分析。
如果实验分组大于两组,你可以将表达矩阵自行拆分,或者自己运行 DESeq2 和DRIMSeq。
输出文件:
运行完成以后输出文件夹(--out_dir)路径下会有以下文件,MCF7和A549是实验分组条件:
genes_deseq2_MCF7_v_A549.tsv基因差异表达矩阵。genes_deseq2_QCplots_MCF7_v_A549.pdfQC 质控图,更多细节参考 DESeq2 manual。isoforms_deseq2_MCF7_v_A549.tsv转录本异构体(isoform)差异表达矩阵。isoforms_deseq2_QCplots_MCF7_v_A549.pdfQC 质控图。isoforms_drimseq_MCF7_v_A549.tsv转录本异构体(isoform)的差异使用矩阵。workdir临时文件,包括过滤掉的输出文件。
选项:
- 必要选项
--counts_matrix Tab-delimited isoform count matrix from flair quantify #flair定量表达矩阵。
--out_dir Output directory for tables and plots. #输出文件夹路径。- 可选选项
--help Show this help message and exit #帮助。
--threads Number of threads for parallel DRIMSeq. #运行DRIMseq的线程数。
--exp_thresh Read count expression threshold. Isoforms in which both
conditions contain fewer than E reads are filtered out (Default E=10) #isoform表达count数阈值,低于此值则舍去。默认值为10。
--out_dir_force Specify this argument to force overwriting of files in
an existing output directory #输出路径。6. flair diffSplice
输入文件:
- 转录本定量表达矩阵:
counts_matrix.tsv。 - isoforms的bed文件:
isoforms.bed
usage: flair_diffSplice -i isoforms.bed -q counts_matrix.tsv [options]这个模块从转录本异构体(isoform)中界定以下四类可变剪切事件(Alternative Splicing,AS):
- intron retention (ir)
- alternative 3’ splicing (alt3)
- alternative 5’ splicing (alt5)
- cassette exons (es)
在可变剪接的所有模式中,外显子跳跃(图4a)是高等真核生物中最常见的可变剪接类型,被跳过的外显子称为盒式外显子(cassette exons)。例如,一个含有A、B、C三个外显子的基因,其最后的mRNA产物有ABC和AC两种,可以被跳过的B外显子就是盒式外显子。
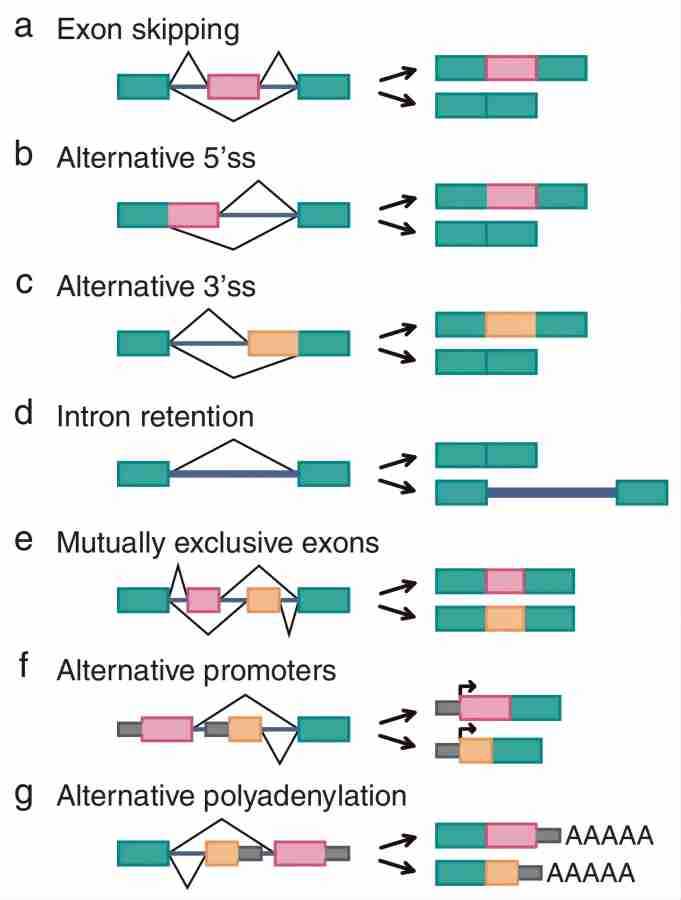
图4. 真核生物中的7种可变剪接模式(图片作者:Choi & Cho)
如果每个分组中的样本数等于或者大于3个,则可以通过--test选项,DRIMSeq将计算两组间的差异可变剪切事件。如果每组没有样本重复,则可以用diffsplice_fishers_exact来进行差异统计学分析。
输出文件:
diffsplice.alt3.events.quant.tsvdiffsplice.alt5.events.quant.tsvdiffsplice.es.events.quant.tsvdiffsplice.ir.events.quant.tsv
如果运行DRIMSeq,这会获得以下结果(A和B为两个分组):
drimseq_alt3_A_v_B.tsvdrimseq_alt5_A_v_B.tsvdrimseq_es_A_v_B.tsvdrimseq_ir_A_v_B.tsvworkdir临时文件,包括过滤掉的输出文件。- 必要选项
--isoforms Isoforms in bed format from Flair collapse. #isoform的bed文件。
--counts_matrix Tab-delimited isoform count matrix from Flair quantify. #isoform表达矩阵
--out_dir Output directory for tables and plots. #输出文件夹路径。- 可选选项
--help Show all options. #帮助选项
--threads Number of processors to use (default 4). #使用线程,默认为4》
--test Run DRIMSeq statistical testing. #使用DRIMSeq进行统计学分析。
--drim1 The minimum number of samples that have coverage over an
AS event inclusion/exclusion for DRIMSeq testing; events
with too few samples are filtered out and not tested (6). #对可变剪切事件(保留和排除)有覆盖度的最小样本数。
--drim2 The minimum number of samples expressing the inclusion of
an AS event; events with too few samples are filtered out
and not tested (3). #包含保留可变剪切事件的最小样本数。
--drim3 The minimum number of reads covering an AS event
inclusion/exclusion for DRIMSeq testing, events with too
few samples are filtered out and not tested (15). #对可变剪切事件(保留和排除)有覆盖度的最小read数。
--drim4 The minimum number of reads covering an AS event inclusion
for DRIMSeq testing, events with too few samples are
filtered out and not tested (5).#包含保留可变剪切事件的最小read数。
--batch If specified with --test, DRIMSeq will perform batch correction. #DRIMSeq可以进行批次校正。
--conditionA Specify one condition corresponding to samples in the
counts_matrix to be compared against condition2; by default,
the first two unique conditions are used. This implies --test. #指定差异分析比对组。
--conditionB Specify another condition corresponding to samples in the
counts_matrix to be compared against conditionA. #指定差异分析比对组。
--out_dir_force Specify this argument to force overwriting of files in an
existing output directory #输出路径。注释:
基因和转录本异构体(isoform)的差异结果根据p值进行筛选和排序,p小于0.05的保留大于0.05的舍去。舍去的结果在workdir文件夹里可以查看。
对于复杂的剪切结果,例如下面所示flair diffSplice结果里2个3'可变剪切,3个内含子保留,和4个外显子跳跃事件,对于每个事件的所有结果,包括保留和去除的转录本异构体:
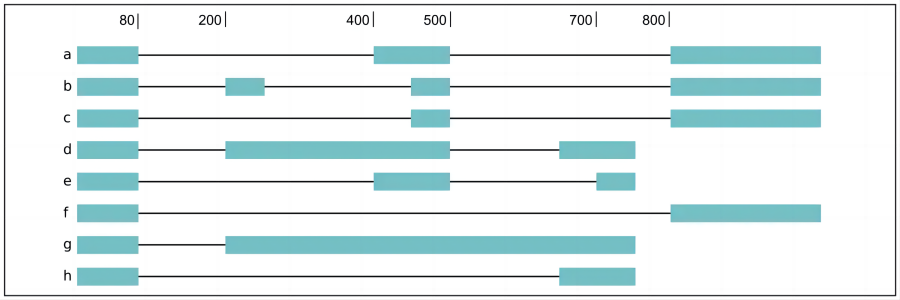
a3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... ea3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... ea3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... e参考文献:
1.Tang, A. D., Soulette, C. M., van Baren, M. J., Hart, K., Hrabeta-Robinson, E., Wu, C. J., & Brooks, A. N. (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nature Communications.
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号