转录组GSE157718_Tpm与Count差异分析的比较
原创
转录组GSE157718_Tpm与Count差异分析的比较
原创
sheldor没耳朵
发布于 2024-08-01 18:56:07
发布于 2024-08-01 18:56:07
转录组GSE157718_Tpm与Count差异分析的比较
在尝试复现GSE157718数据集的时候,发现网站同时提供了表达矩阵tpm形式与count形式,因此分别用这两种形式进行基因差异与富集分析,再进行对比。
注:有count矩阵就用count矩阵
1 Count形式
以count给出的表达矩阵是我们最为熟悉的形式,这里只稍加记录下数据整理的代码,具体的差异富集分析,与其他的流程并无不同。
1 以fread函数导入的数据形式为data.table,设置行名很麻烦,这里先转化为data.frame形式
2 行名或(GeneID列)为ENTREZID,需要转化为SYMBOL
3 归根结底是表达矩阵的形式需要行名为基因名,列为数据集,所有的操作往这个方向努力就行
表达矩阵exp
library(data.table)
library(tinyarray)
dat = fread("GSE157718_raw_counts_GRCh38.p13_NCBI.tsv.gz")
dim(dat)
#> [1] 39376 7
colnames(dat)
#> [1] "GeneID" "GSM4773688" "GSM4773689" "GSM4773690" "GSM4773691"
#> [6] "GSM4773692" "GSM4773693"
#data.table转化为data.frame
dat <- as.data.frame(dat)
class(dat)
#> [1] "data.frame"
#ENTREZID转化为SYMBOL
library(org.Hs.eg.db)
library(clusterProfiler)
output <- bitr(dat$GeneID,
fromType = 'ENTREZID',
toType = 'SYMBOL',
OrgDb = 'org.Hs.eg.db')
#> Warning in bitr(dat$GeneID, fromType = "ENTREZID", toType = "SYMBOL", OrgDb =
#> "org.Hs.eg.db"): 4.28% of input gene IDs are fail to map...
colnames(output)
#> [1] "ENTREZID" "SYMBOL"
exp <- merge(dat, output, by.x = "GeneID", by.y = "ENTREZID")
exp <- exp[!duplicated(exp$SYMBOL),]
rownames(exp) <- exp$SYMBOL
exp <- exp[,c(-1,-8)]
exp = as.matrix(exp)基因过滤与分组信息
#基因过滤
#分组信息
colnames(exp) <- c("NS1","NS2","NS3","ES1","ES2","ES3")
library(stringr)
Group = ifelse(str_detect(colnames(exp),"ES"),"ES","NS")
Group = factor(Group,levels = c("ES","NS"))
table(Group)
#> Group
#> ES NS
#> 3 3
data.frame(colnames(exp),Group)
#> colnames.exp. Group
#> 1 NS1 NS
#> 2 NS2 NS
#> 3 NS3 NS
#> 4 ES1 ES
#> 5 ES2 ES
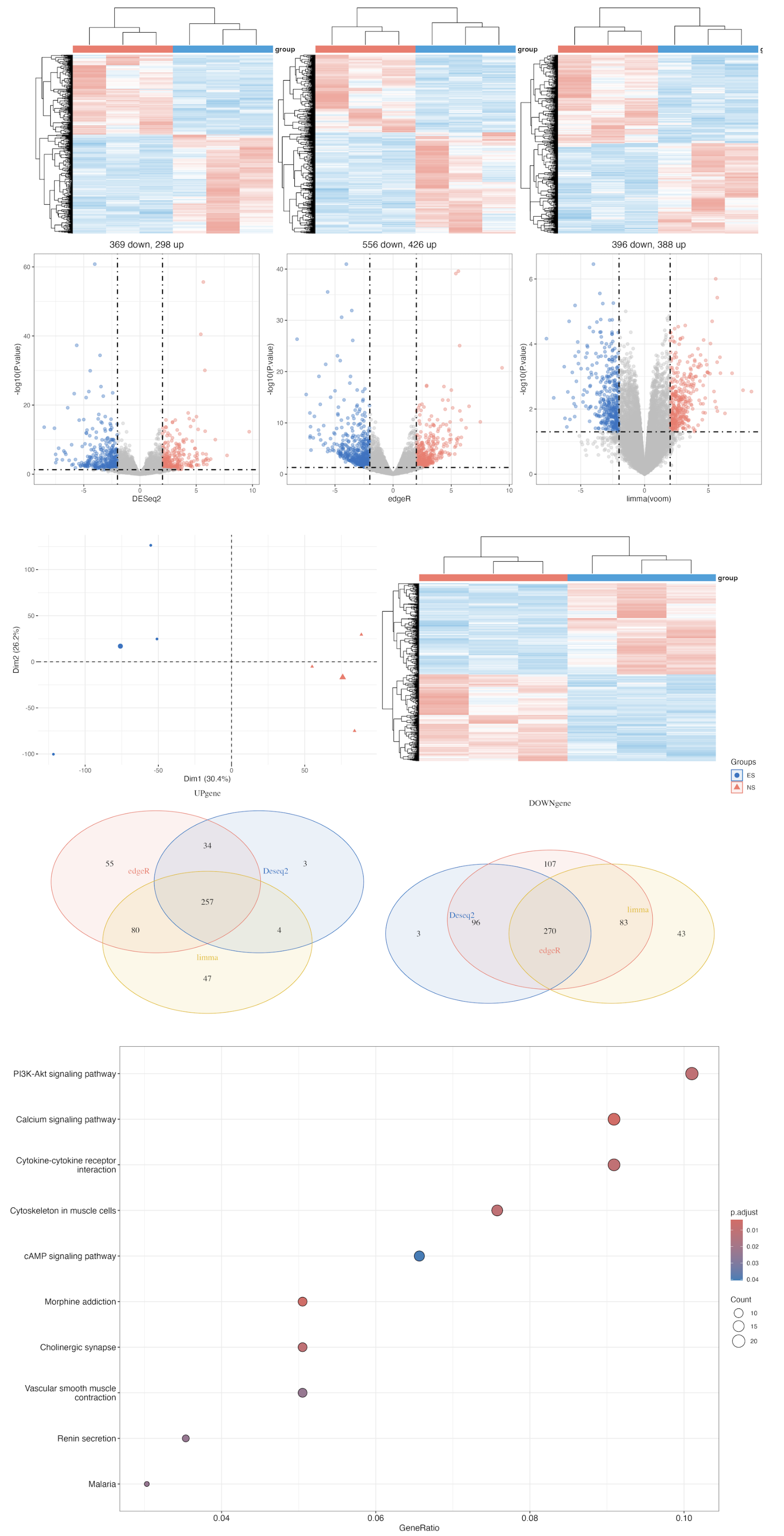
#> 6 ES3 ES以logFC_t = 2,pvalue_t = 0.05为阈值,以DEseq2,edgeR,limma三个R包分别进行差异分析,最好再去交集进行富集分析的结果如下
2 Tpm形式

Tpm也可以勉强进行差异分析,但是只能取log后,用limma做差异分析
fpkm、rpkm需先转换为Tpm形式,用limma做差异分析
limma差异分析参考基于芯片的分析流程
表达矩阵exp
这次需要将ENSEMBL转换为SYMBOL
proj = "GSE157718"
library(data.table)
library(tinyarray)
dat = fread("GSE157718_gene_tpm_matrix.txt")
#data.table转化为data.frame
dat <- as.data.frame(dat)
class(dat)
#> [1] "data.frame"
rownames(dat) <- dat$gene_id
dat <- dat[,-1]
exp = as.matrix(dat)
exp = trans_exp_new(exp,species = "human")
#> Warning in AnnoProbe::annoGene(rownames(exp), ID_type = "ENSEMBL", species =
#> species): 0.13% of input IDs are fail to annotate...基因过滤与分组信息
重点是基因过滤后(或之前)执行exp <- log(exp+1)
!!!!
nrow(exp)
#> [1] 60512
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
#> [1] 20938
range(exp)
#> [1] 0.00 35719.68
exp <- log(exp+1)
#分组信息
colnames(exp)
#> [1] "ES1" "ES2" "ES3" "NS1" "NS2" "NS3"
library(stringr)
Group = ifelse(str_detect(colnames(exp),"ES"),"ES","NS")
Group = factor(Group,levels = c("ES","NS"))
table(Group)
#> Group
#> ES NS
#> 3 3
data.frame(colnames(exp),Group)
#> colnames.exp. Group
#> 1 ES1 ES
#> 2 ES2 ES
#> 3 ES3 ES
#> 4 NS1 NS
#> 5 NS2 NS
#> 6 NS3 NS基因差异分析与可视化
用limma包分析类似基于芯片平台的分析
rm(list = ls())
load("GSE157718.Rdata")
table(Group)
#> Group
#> ES NS
#> 3 3
range(exp)
#> [1] 0.00000 10.48349
# 使用limma包进行差异分析
library(limma)
library(dplyr)
design = model.matrix(~Group)
fit = lmFit(exp,design)
fit = eBayes(fit)
deg = topTable(fit,coef = 2,number = Inf)
# 使用阈值
logFC_t = 1
p_t = 0.05
k1 = (deg$P.Value < p_t)&(deg$logFC < -logFC_t)
k2 = (deg$P.Value < p_t)&(deg$logFC > logFC_t)
deg = mutate(deg,change = ifelse(k1,"Down",ifelse(k2,"Up","Not")))
#Down Not Up
# 158 20605 175
table(deg$change)
#>
#> Down Not Up
#> 158 20605 175
DEG = mutate(deg,symbol = rownames(deg))
save(DEG,Group,file = paste0(proj,"_tpm_DEG.Rdata"))
#可视化
library(ggplot2)
this_title <- paste0('Threshold of logFC is 1',
'\nThe number of up gene is ',nrow(deg[deg$change == 'Up',]) ,
'\nThe number of down gene is ',nrow(deg[deg$change == 'Down',])
)
p1 <- ggplot(data = DEG, aes(x = logFC, y = -log10(P.Value))) +
geom_point(alpha=0.4, size=3.5, aes(color=change)) +
scale_color_manual(values=c("blue", "grey","red"))+
geom_hline(yintercept = -log10(p_t),lty=4,col="black",linewidth=0.8) +
theme_bw()+
ggtitle(this_title )+
theme(panel.grid = element_blank(),
plot.title = element_text(size=8,hjust = 0.5),
legend.title = element_blank(),
legend.text = element_text(size=8))
ggsave("DEG_tpm.png",plot = p1,height = 15,width = 13)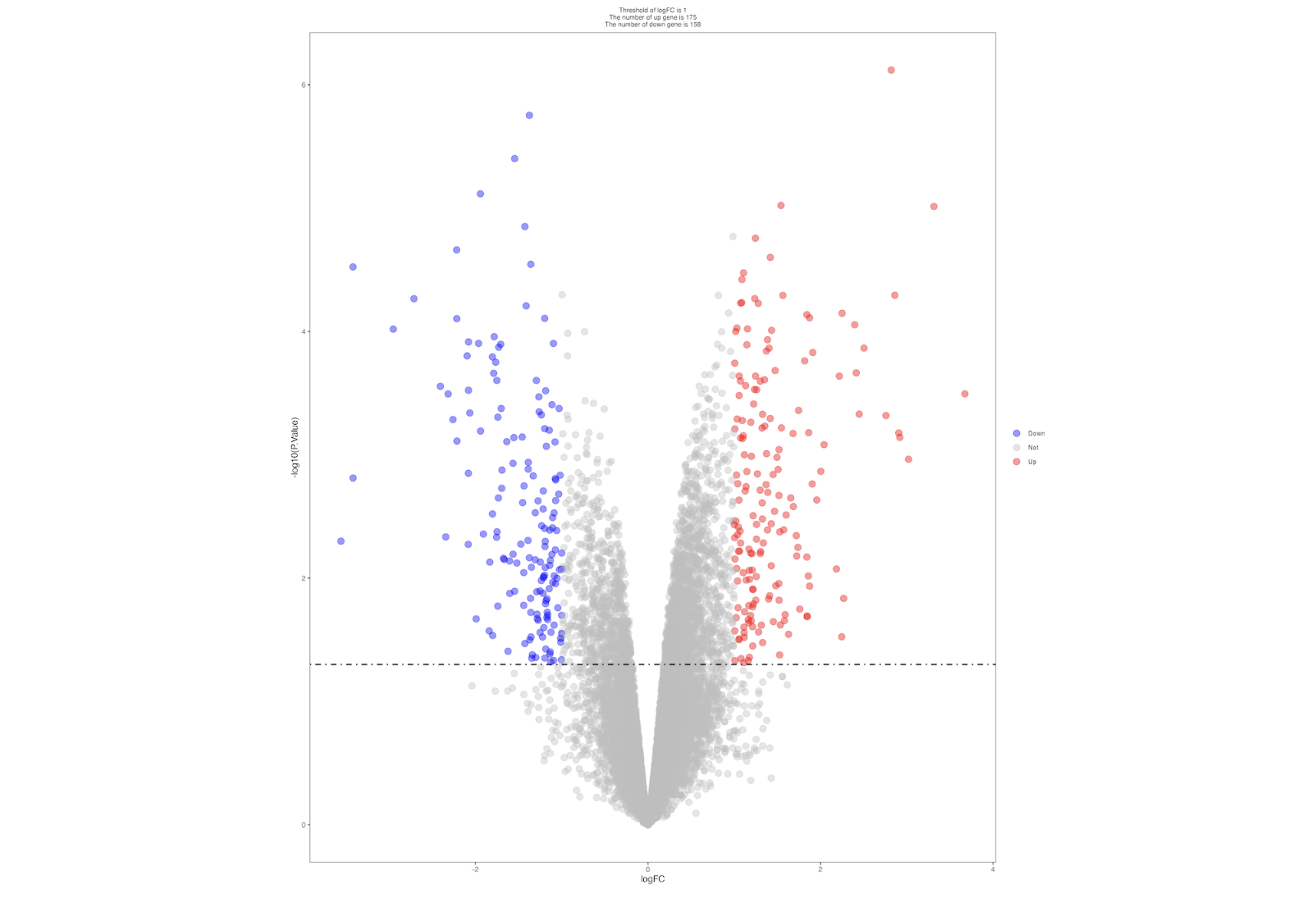
富集分析
rm(list = ls())
library(clusterProfiler)
library(ggthemes)
library(org.Hs.eg.db)
library(dplyr)
library(ggplot2)
library(stringr)
library(enrichplot)
#(1)输入数据
load("GSE157718_tpm_DEG.Rdata")
library(tinyarray)
g <- rownames(DEG)[DEG$change!="Not"]
output <- bitr(g,
fromType = 'SYMBOL',
toType = 'ENTREZID',
OrgDb = 'org.Hs.eg.db')
gene_diff = output$ENTREZID
#(2)富集
ekk <- enrichKEGG(gene = gene_diff,
organism = 'hsa')
ekk <- setReadable(ekk,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID")
dotplot(ekk)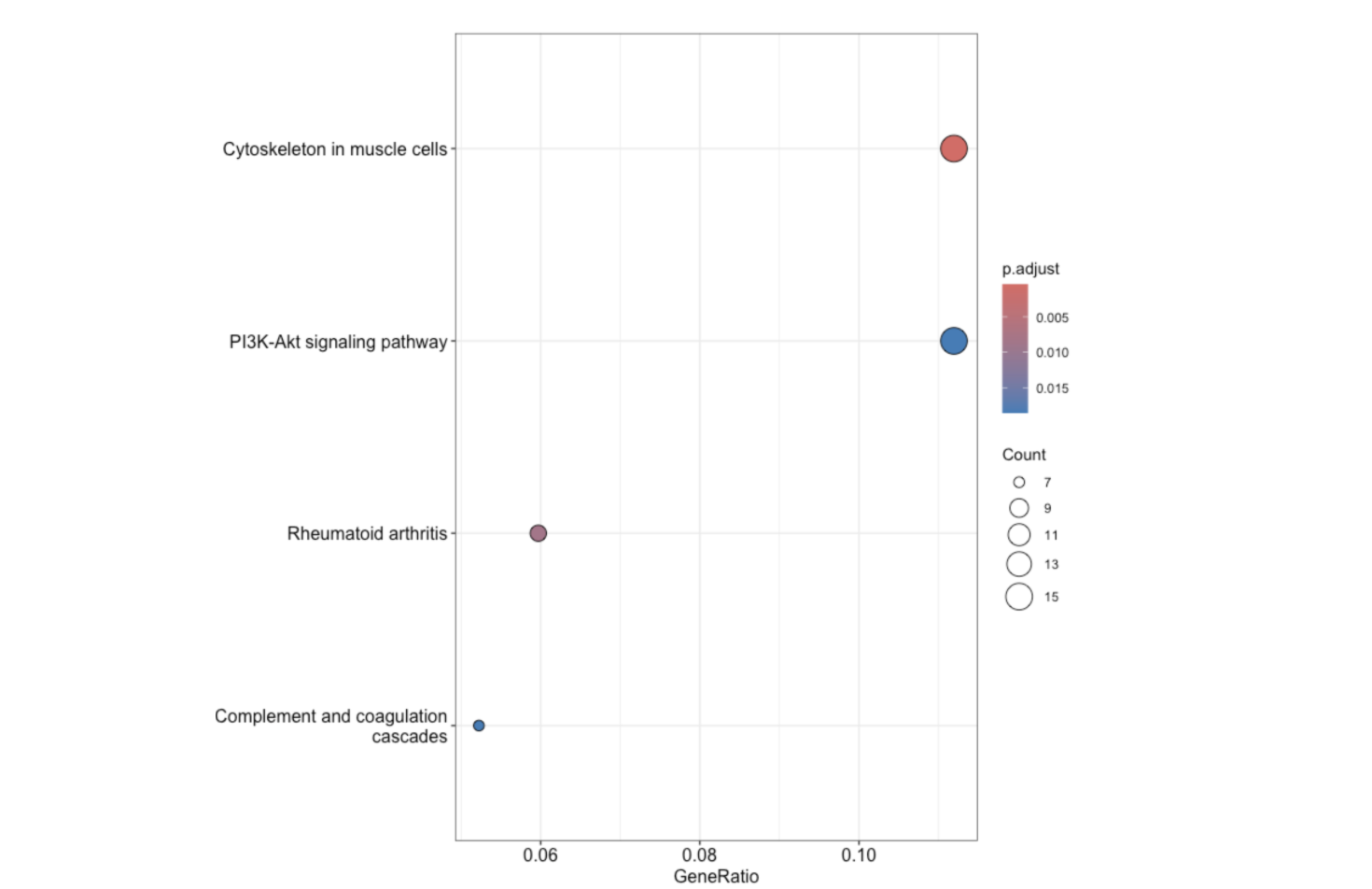
3 Count形式 Vs Tpm形式
rm(list = ls())
library(data.table)
load("GSE157718_count_DEG.Rdata")
#DEG3为limma包分析的结果
DEG_count <- DEG3
rm(DEG1);rm(DEG2);rm(DEG3)
load("GSE157718_tpm_DEG.Rdata")
DEG_tpm <- DEG;
rm(DEG)
ids=intersect(rownames(DEG_count),
rownames(DEG_tpm))
df1= data.frame(
DEG_count1 = DEG_count[ids,"logFC"],
DEG_tpm1 = DEG_tpm[ids,"logFC"]
)
library(ggpubr)
comp_between_DEG1 <- ggscatter(df1, x = "DEG_count1", y = "DEG_tpm1",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
ggsave("comp_between_count_tpm.png",width = 15,height = 13)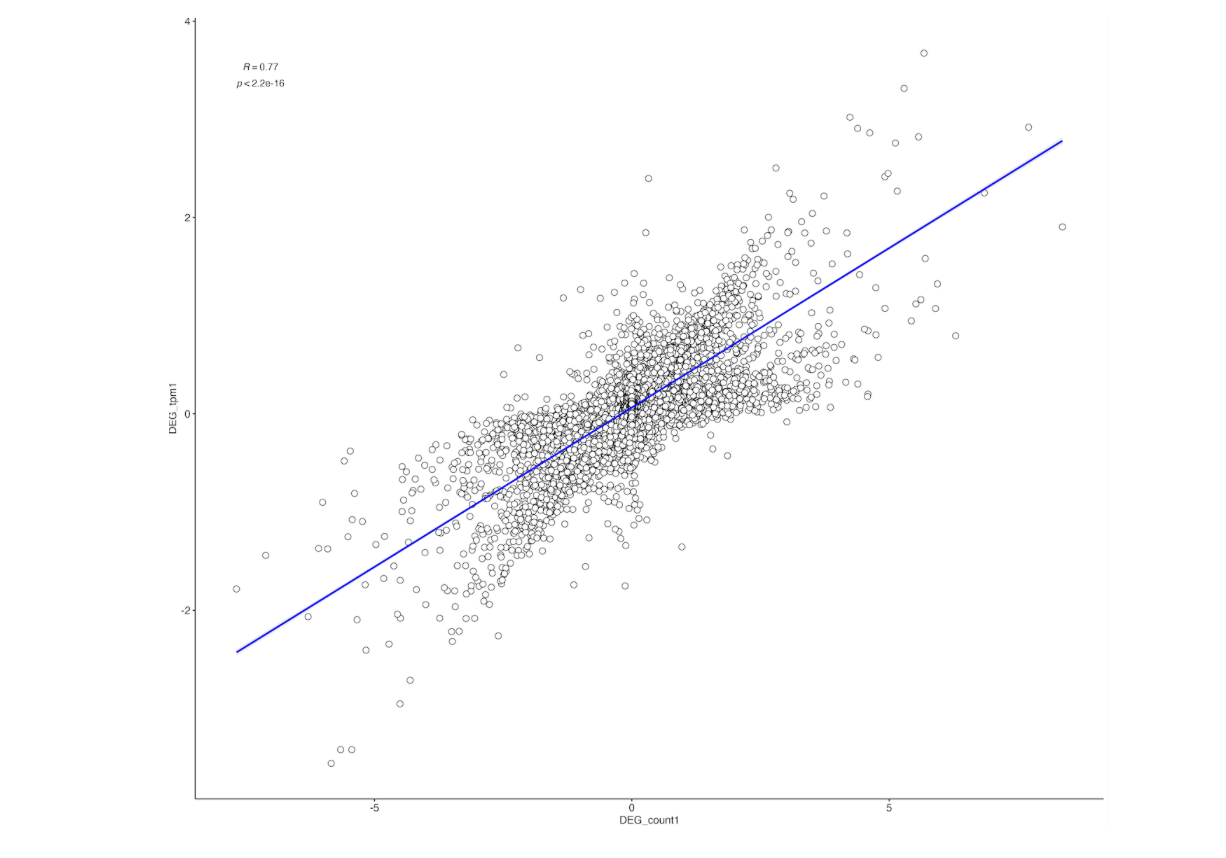
df2= data.frame(
DEG_count2 = DEG_count[ids,"change"],
DEG_tpm2 = DEG_tpm[ids,"change"]
)
p2 <- gplots::balloonplot(table(df2))
ggsave("balloon.png",plot = p2,width = 15,height = 13)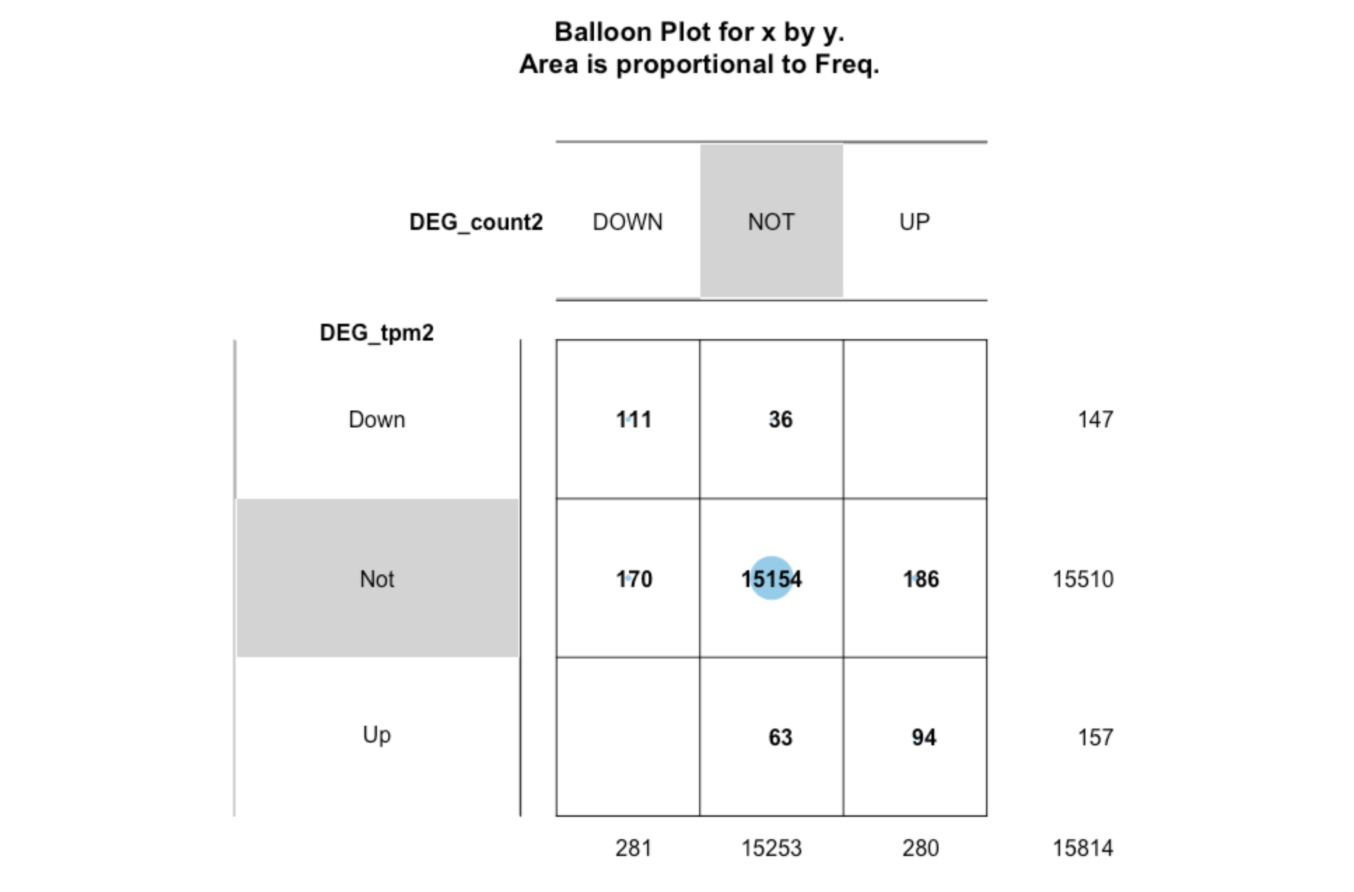
symbols_list = split(ids,paste(df2[,1],df2[,2]))
library(clusterProfiler)
library(org.Hs.eg.db)
library(ReactomePA)
library(ggplot2)
library(stringr)
# 首先全部的symbol 需要转为 entrezID
gcSample = lapply(symbols_list, function(y){
y=as.character(na.omit(AnnotationDbi::select(org.Hs.eg.db,
keys = y,
columns = 'ENTREZID',
keytype = 'SYMBOL')[,2])
)
y
})
#gcSample
pro='comp'
# 第1个注释是 KEGG
xx <- compareCluster(gcSample, fun="enrichKEGG",
organism="hsa", pvalueCutoff=0.3)
dotplot(xx) + theme(axis.text.x=element_text(angle=45,hjust = 1)) +
scale_y_discrete(labels=function(x) str_wrap(x, width=50))
ggsave(paste0(pro,'comp_kegg.pdf'),width = 10,height = 8)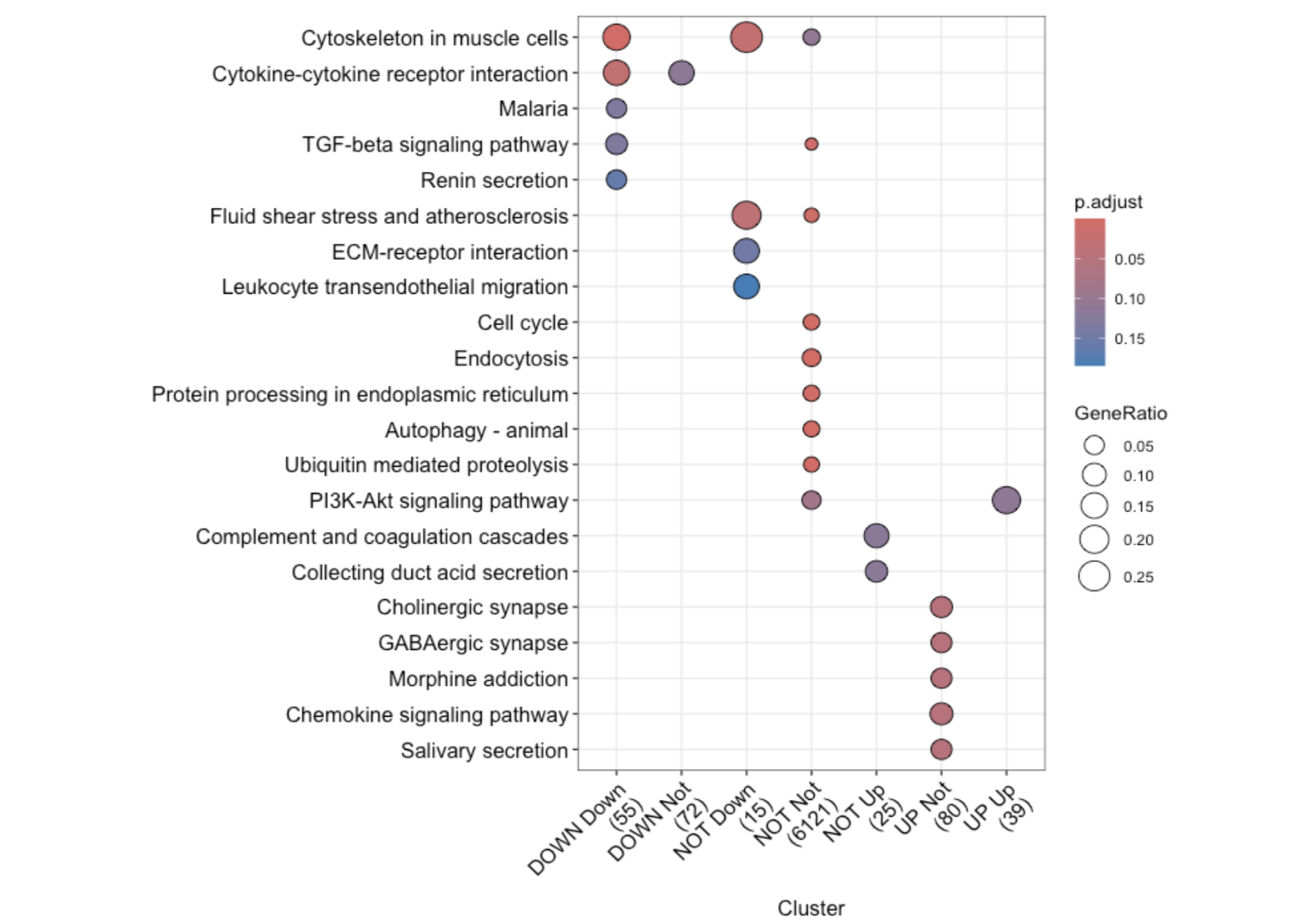
由此可见,同一个数据集采用Count与Tpm形式做出来的差异与富集分析结果还是有较大差别的,这里的Tpm logFC的阈值为1(设置为2的话分析出来的差异基因只有30左右),同Count 的logFC的阈值为2相比,富集的通路类型反而少了很多。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号