R语言基础| 下载、安装

R语言基础| 下载、安装

生物信息学需要复杂的统计学分析和灵活的数据可视化。编程语言R拥有丰富的统计学函数和数据可视化包,适用于高维生物学数据。基于此,本系列文章开展R语言基础教程,帮助更多学习生信的小伙伴打好编程基础。
R下载与安装
下载链接:https://mirrors.dftianyi.com/CRAN/
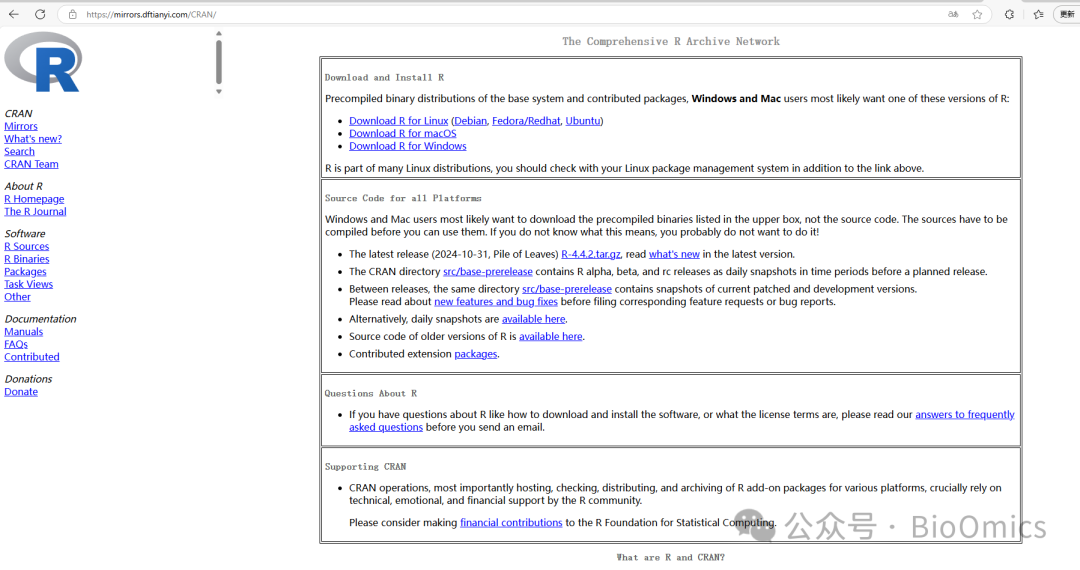
根据电脑系统选择相应的R版本
- mac系统的根据macOS版本号选择合适的安装包,下载并安装即可。

- windows系统的按以下步骤下载并安装即可
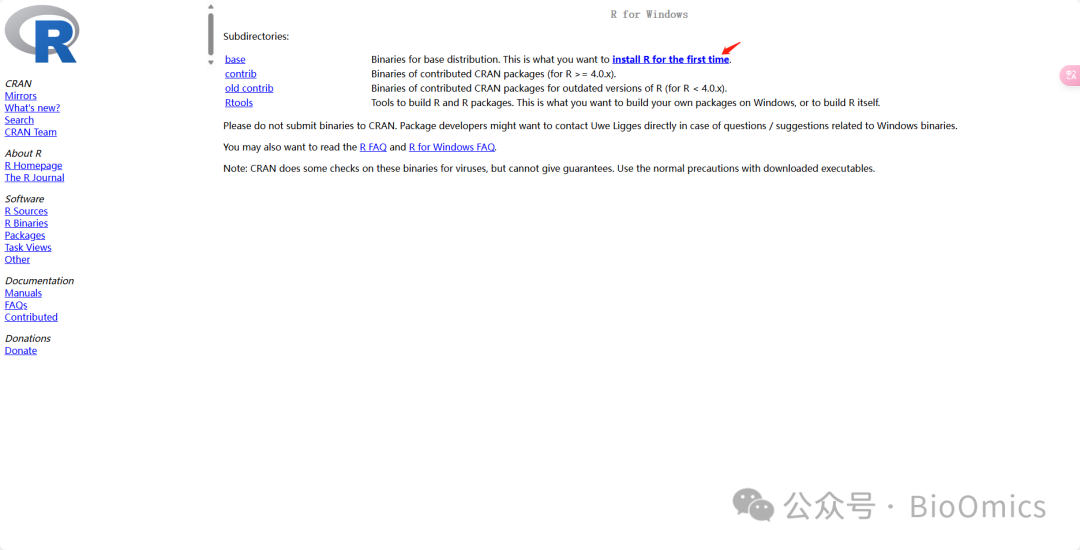
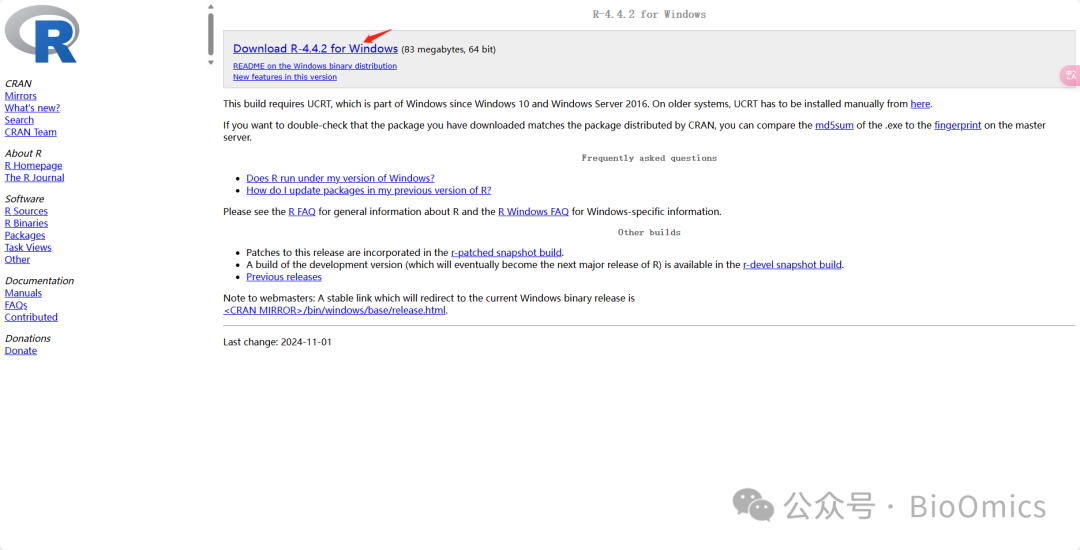
- Linux系统安装R
使用Linux安装R,建议配置好conda环境之后,用conda命令安装R,之后有需要安装的R包,conda会解决大部分的包依赖问题。
搭建分析环境
conda是一个包和环境管理工具,用于创建、隔离环境,避免环境冲突。
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh安装Miniconda之后,通过命令行创建、列出、激活、删除环境,以及安装和卸载包。
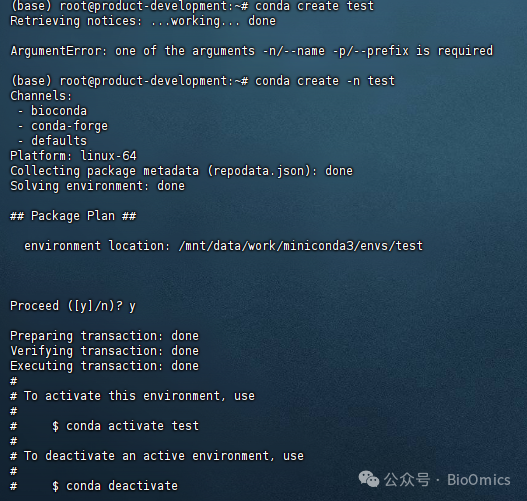
激活conda环境之后,运行R,没有安装会给出提示
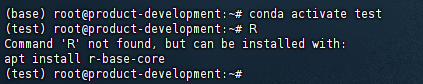
安装之前可以先查找一下R有哪些版本,根据需要选择合适的版本安装。
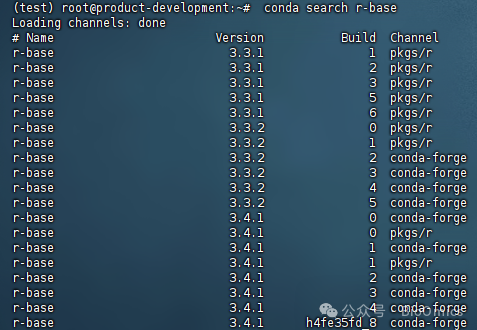
conda install r-base=4.4.1除了conda安装还可以:
sudo apt install r-base # 如果没有管理员权限无法操作
# 这样装上R默认在/usr/bin/RRstudio-server的安装与配置
RStudio是一款集成开发环境(IDE)。R自带的环境操作起来可能不是很方便,而Rstudio很好地解决了这个问题,且具有调试、可视化等功能,支持纯R脚本、Rmarkdown、Bookdown、Shiny等。
下载链接:https://posit.co/download/rstudio-desktop/
Linux安装Rstudio-sever:
- 安装Rstudio-sever:
#安装编译器:
sudo apt-get install gdebi-core # 如果没有管理员权限无法操作
# 下载安装包:
wget https://download2.rstudio.org/server/bionic/amd64/rstudio-server-2021.09.0-351-amd64.deb
# 安装Rstudio-server:
sudo gdebi rstudio-server-2021.09.0-351-amd64.deb # 如果没有管理员权限无法操作
- 配置Rstudio-sever:
# 修改配置文件:
echo 'www-port=你的端口号' >> /etc/rstudio/rserver.conf# 具体的端口号可以问你的管理员
# 重启rstudio使配置生效
sudo rstudio-server restart
# 查看Rstudio-server运行状态:
sudo rstudio-server status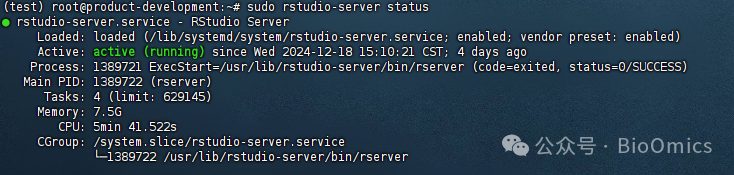
之后就可以在浏览器中通过IP:端口的形式访问Rstudio-sever
- 为Rstudio-server创建新登录用户
Linux中的Rstudio-server无法像Windows那样用一个用户就可以开启多个Rstudio,实际使用中Rstudio-server的一个用户只能使用一个Rstudio窗口,若需要同时开启多个Rstudio窗口,可启用子账号:
# 创建一个新用户,并且指定其目录:
useradd -d /home/新用户名 -m 新用户名
# 然后设置密码:
passwd 需要修改密码的用户名
# 新建的用户未指定shell。我们只需将其指定为/bin/bash重新登录即可。否则该用户命令行只有一个$
usermod -s /bin/bash 新用户名完成以上操作就可以用新用户登录Rstudio了
R的使用
- 用于管理R工作区的函数
函数 | 功能 |
|---|---|
getwd() | 显示当前的工作目录 |
setwd(“路径”) | 设置或修改当前的工作目录 |
Is() | 列出当前工作区中的对象 |
rm(objectlist) | 移除(删除)一个或多个对象 |
help(options) | 显示可用选项的说明 |
options() | 显示或设置当前选项 |
save.image(“myfile”) | 保存工作区到文件myfile中(默认值为.RData) |
save(objectlist,file=“myfile”) | 保存指定对象到一个文件中 |
load(“myfile”) | 读取一个工作区到当前会话中(默认值为.RData) |
dir.create() | 创建新目录 |
- R包的安装和使用
包的基本函数
这里涉及需要包名的地方用Seurat举例,实操中需要其他包则改为具体的包名;
函数 | 操作 |
|---|---|
.libPaths() | 显示库(库即存储包的目录)所在的位置 |
library() | 显示库中有哪些包 |
search() | 显示哪些包已被加载并可以使用 |
install.packages(“Seurat”) | 安装Seurat包,注意双引号必须有 |
update.packages(“Seurat”) | 更新包,注意双引号必须有 |
library(Seurat) | 加载包,无需引号 |
installed.packages() | 列出已安装的包 |
Seurat使用示例展示
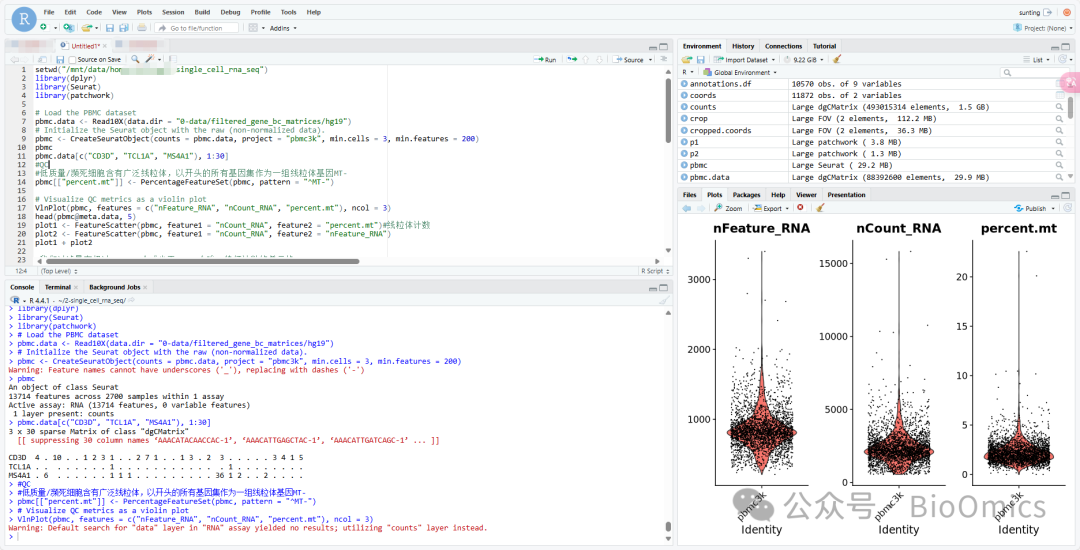
代码:
setwd("/mnt/data/home/xxx/2-single_cell_rna_seq")
library(dplyr)
library(Seurat)
library(patchwork)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "0-data/filtered_gene_bc_matrices/hg19")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
#QC
#低质量/濒死细胞含有广泛线粒体,以开头的所有基因集作为一组线粒体基因MT-
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
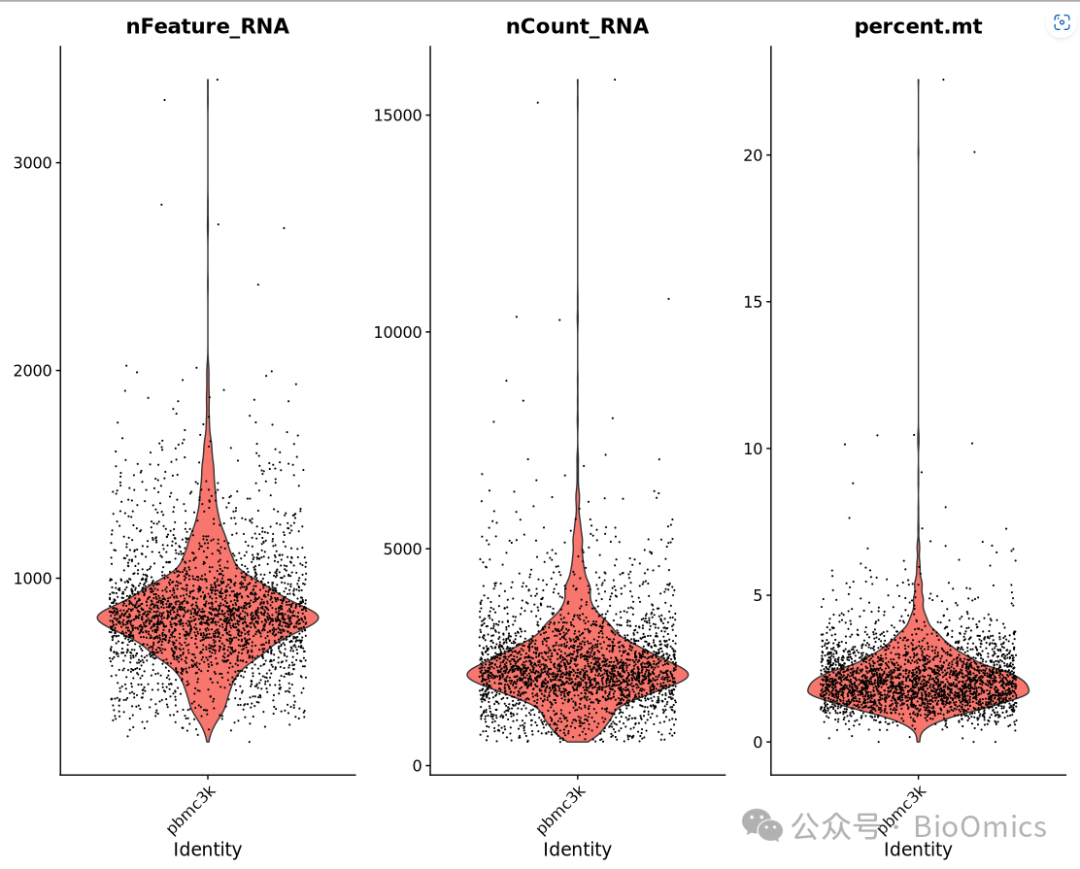
# Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)结果图:
下期内容
下一节我们将详细介绍"R语言管理数据集"的内容
腾讯云开发者
