知识分享--肿瘤相关成纤维细胞在肿瘤耐药和肿瘤进展中的作用
原创
知识分享--肿瘤相关成纤维细胞在肿瘤耐药和肿瘤进展中的作用
原创
追风少年i
发布于 2025-07-28 11:57:22
发布于 2025-07-28 11:57:22
作者,Evil Genius
今天我们分享一些知识。
关于肿瘤环境中的成纤维细胞,王凌华的文章可谓一颗“炸弹”。


nature期刊也进行了重点解读,可见其重要性。
今天我们来总结一下肿瘤相关成纤维细胞在肿瘤耐药和肿瘤进展中的作用
在肿瘤微环境中,CAFs通过以下机制发挥作用:分泌ECM成分以稳定组织结构、通过降解作用重塑ECM、诱导上皮-间质转化促进转移,以及抑制癌症免疫应答。
CAFs通过独特机制(包括分泌ECM构建药物渗透物理屏障、抑制抗肿瘤免疫、促进血管生成及增强癌细胞迁移)导致治疗抵抗和预后不良。
肿瘤微环境(TME)
肿瘤组织中存在包括免疫细胞、成纤维细胞和血管内皮细胞在内的多种非癌细胞。这些宿主源性细胞在肿瘤生长中发挥关键作用,且其特性与正常宿主细胞存在显著差异。CAFs通过与免疫细胞、血管内皮细胞等相互作用调控TME,在其中居于核心地位。
TME中的成纤维细胞
在开放性伤口中,成纤维细胞会被激活为肌成纤维细胞,通过分泌胶原蛋白促进愈合。此过程会表达α-平滑肌肌动蛋白(SMA)和成纤维细胞活化蛋白(FAP)等激活标志物。伤口愈合完成后,活化的成纤维细胞会恢复静息状态。但正如"肿瘤是不愈合的伤口"这一经典论断所示,肿瘤组织中的成纤维细胞活化对癌症进展至关重要。TME中的成纤维细胞活化程度远超正常伤口愈合——它们会转化为CAFs并过度产生基质成分。此外,在炎症因子、局部缺氧和CAF外泌体作用下,星状细胞、内皮细胞、周细胞、脂肪细胞及间皮细胞也可分化为CAFs。
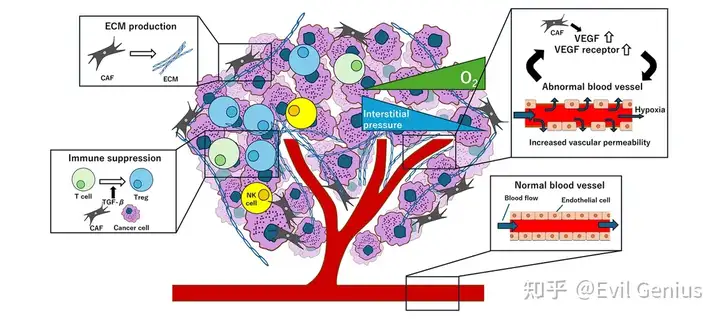
肿瘤进展早期,癌细胞分泌的IL-1β等细胞因子可通过激活NF-κB通路,诱导正常成纤维细胞分化为促炎性CAFs。类似地,缺氧环境会使与肿瘤类器官共培养的胰腺星状细胞分化为具有炎症表型的CAFs。CAF外泌体中的miRNA同样能促使正常成纤维细胞转化。由于表观遗传修饰的影响,这些转化具有不可逆性,CAFs几乎不可能恢复为静息状态。CAFs这种独特性质与其起源和活化过程密切相关:其在TME中的异常活化及多样化的细胞来源(不仅限于成纤维细胞),造就了高度异质性的特性。
TME中的免疫细胞
TME中存在T细胞、巨噬细胞、自然杀伤(NK)细胞等多种免疫细胞,它们本应发挥抗肿瘤免疫作用。这种免疫应答通常针对肿瘤新抗原。但CAFs会重塑免疫格局以促进肿瘤生长:例如通过缺乏共刺激分子的抗原呈递诱导T细胞分化为调节性T细胞(Tregs);或通过分泌细胞因子/趋化因子促使M2型巨噬细胞和髓源性抑制细胞(MDSCs)迁移。这些机制导致治疗抵抗(尤其是癌症免疫疗法)和不良预后。
TME中的血管系统
失控增殖的癌细胞需要血液供应,因此TME中常诱导产生促血管生成的血管内皮生长因子(VEGF)。CAFs除分泌VEGF外,还能通过STAT3信号通路(不依赖VEGF)诱导血管生成。但肿瘤血管内皮呈不连续状,与正常组织血管显著不同,导致整个TME处于高间质压力和缺氧状态——这种环境既支持肿瘤生长,又促进治疗抵抗的形成。
肿瘤微环境中的细胞外基质(ECM)
ECM是所有器官和组织中存在的非细胞成分,主要包含胶原蛋白、蛋白聚糖和弹性蛋白等。在肿瘤微环境(TME)中,ECM通过持续的生成与降解进行动态重塑。如前述,癌症相关成纤维细胞(CAFs)通过分泌基质金属蛋白酶(MMPs)参与ECM的生成与降解过程。
ECM不仅为癌组织提供机械支撑,还具有以下关键功能:
生长因子储存库:在特定条件下向癌细胞释放生长因子
信号传导支架:促进癌细胞与生长因子的相互作用
动态调控平台:通过重塑(降解)作用:
✓ 调节支架结构与癌细胞的相互作用 ✓ 调控细胞代谢 ✓ 影响多种信号通路
CAFs亚型分类及其在肿瘤生长中的作用
单细胞分析揭示CAFs并非均质细胞群。虽然其主要功能是产生ECM成分,但CAFs还参与多种其他功能,这些功能既可能促进也可能抑制肿瘤生长。本节将探讨不同CAF亚型及其在肿瘤生长中的作用。
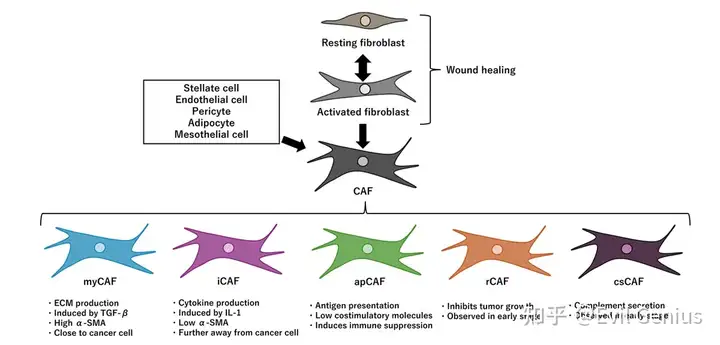
肌成纤维样CAFs(myCAFs)
myCAFs在ECM生成中起关键作用。该亚型高表达肌成纤维细胞标志物α-SMA,具有参与伤口愈合的典型特征,主要功能是产生ECM。myCAFs邻近肿瘤细胞分布,对TGF-β高度敏感——TGF-β可促进祖细胞分化为myCAFs,导致ECM过度生成。研究推测,myCAFs与癌细胞的近距离分布可通过ECM形成的物理屏障有效促进治疗抵抗。临床数据显示,肿瘤组织中myCAFs高比例与ECM富集及不良预后相关。但矛盾的是,myCAFs也可能部分抑制肿瘤生长:例如肌成纤维细胞缺失会导致小鼠胰腺癌恶化并促进结直肠癌转移;而myCAFs中I型胶原缺失会通过MDSCs迁移加速肿瘤进展并抑制癌症免疫。
炎症性CAFs(iCAFs)
iCAFs通过JAK/STAT和NF-κB信号通路产生CXCL-1、IL-1、IL-6、基质细胞衍生因子-1、肝细胞生长因子及CCL17等炎症因子。这些细胞因子可诱导MDSCs、M2巨噬细胞和Tregs向肿瘤迁移并激活,从而抑制抗癌免疫反应。iCAFs高表达IL-1受体而低表达α-SMA,由IL-1诱导产生且远离癌细胞分布。值得注意的是,iCAFs与myCAFs存在相互转化关系,可响应IL-1和TGF-β等细胞因子发生表型转换。虽然iCAFs与癌细胞空间分布的功能意义尚不明确,但这种分布模式可能有利于肿瘤生长——iCAFs位于myCAFs构建的ECM物理屏障外围,可高效分泌细胞因子来招募Tregs等免疫抑制细胞。
抗原呈递CAFs(apCAFs)
apCAFs特征性表达MHC II类分子和CD74,但缺乏CD80/CD86/CD40等共刺激分子,因此无法像专业抗原呈递细胞那样激活免疫功能。该亚型能以抗原特异性方式直接诱导初始CD4+ T细胞分化为Tregs。在PDAC中,apCAFs由间皮细胞通过IL-1和TGF-β介导转化而来(伴随间皮特征丢失和成纤维特征获得)。关于空间分布,乳腺癌TME中的apCAFs可能不像iCAFs或myCAFs具有特定分布模式。
最新研究发现,某些apCAFs可激活CD4+ T细胞:在人肺肿瘤组织中观察到被CD4+ T细胞(非Tregs)环绕的apCAFs,这些细胞通过MHC II类分子呈递抗原激活CD4+ T细胞。这表明即使相同分子亚型,CAFs在不同癌种中可能具有功能异质性。
肿瘤抑制性CAFs(rCAFs)
rCAFs是CAFs中抑制肿瘤生长的特殊亚型,主要存在于肿瘤组织早期阶段,随着肿瘤进展逐渐减少。目前尚无其空间分布报道,这可能与肿瘤进展后rCAFs难以鉴定有关。
rCAFs具有与正常成纤维细胞相似的抑癌特性:
- Meflin(正常成纤维细胞标志物):通过抑制赖氨酰氧化酶(介导胶原交联)降低ECM胶原含量,改善组织软化和药物渗透。胰腺癌患者中meflin高表达者术后生存期显著延长
- Asporin(正常成纤维细胞标志物):通过结合TGF-β1抑制TGF-β/Smad通路。乳腺癌患者asporin高表达与较好预后相关,但前列腺癌模型显示asporin缺失可减少肿瘤干细胞和间质细胞,增加CD8+细胞——这种矛盾结果提示asporin作用可能因癌种和TME状态而异
补体分泌CAFs(csCAFs)
仅存在于早期PDAC的新型CAF亚型。csCAFs主要分泌C3、C7等补体蛋白,其具体功能和空间分布尚未阐明。值得注意的是,PDAC新辅助治疗中,CAFs分泌的补体蛋白表达增强与患者生存改善相关,提示csCAFs可能影响癌症治疗效果。
CAFs介导肿瘤进展与治疗抵抗的机制
CAFs通过产生胶原等ECM成分,在细胞因子分泌、抗肿瘤免疫抑制及肿瘤生长调控中发挥多重作用。
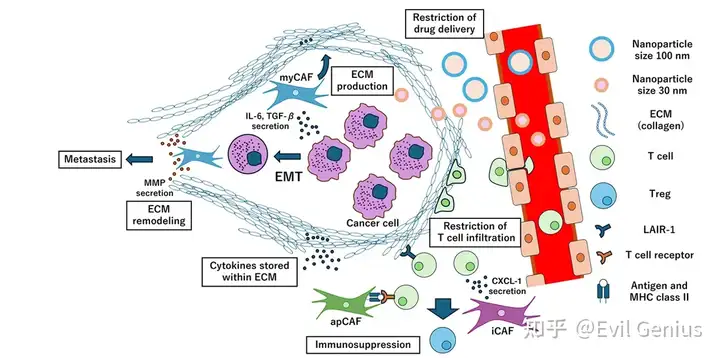
ECM生成在肿瘤进展与治疗抵抗中的作用
活化成纤维细胞的核心功能是产生ECM,而CAFs则处于持续活化状态。过量ECM对癌症治疗构成多重障碍:
1. 削弱EPR效应限制药物渗透
肿瘤血管的高通透性和淋巴回流缺失本应通过EPR效应促进药物渗透,但在PDAC和乳腺癌等ECM富集型肿瘤中该效应显著受限。临床数据显示:
- 脂质体阿霉素虽能减少转移性乳腺癌不良反应,但无进展生存期未见显著改善
- 多项临床试验荟萃分析证实:脂质体化疗与传统化疗疗效无显著差异
这种治疗瓶颈与纳米颗粒尺寸直接相关:
✓ 肿瘤血管可允许100nm颗粒通过 ✓ 但ECM富集肿瘤核心区仅允许≤30nm颗粒有效渗透 ✓ 常规脂质体制剂直径约100nm,导致其穿透血管后仍无法深入肿瘤组织
2. 高间质压力与ECM阻碍T细胞浸润
- 高间质压力抑制T细胞从血管外渗
- 致密ECM网格结构阻碍T细胞的阿米巴样运动(该运动不依赖黏附分子)
- ECM物理性减少T细胞迁移空间,影响其与肿瘤细胞接触,最终削弱抗肿瘤免疫应答 这一机制是癌症免疫治疗抵抗的重要风险因素
3. ECM结构域直接抑制免疫细胞功能
- T细胞/NK细胞表达的LAIR-1受体与胶原结构域结合后,可激活抑制性信号通路
- 纤维连接蛋白与LILRB4受体的相互作用具有类似免疫抑制效果
4. 作为生长因子储库与力学信号调控者
ECM通过LTBP蛋白储存TGF-β前体(LAP-TGF-β复合物),在降解时释放活性TGF-β
乳腺癌组织弹性模量可达正常组织的4倍(4000 Pa vs 1000 Pa):
✓ ECM硬度促进癌细胞干性 ✓ 过度硬化反而诱导癌细胞休眠 ✓ 结直肠癌肝转移中,ECM力学作用促使肝星状细胞分泌FFAs,癌细胞通过摄取FFAs获得治疗抵抗
诱导癌细胞迁移的机制
CAFs通过两种主要途径促进远端转移:
1. 诱导上皮-间质转化(EMT)
- CAFs分泌TNF-α/IL-6/IL-1β/TGF-β等细胞因子
- iCAFs旁分泌的IL-6可促进膀胱癌细胞EMT,增强其增殖、迁移和侵袭能力
- 尽管直接证据有限,但鉴于iCAFs分泌多种EMT诱导因子,其促转移潜力值得关注
2. ECM重塑介导的转移
- CAFs分泌MMPs重塑ECM,为癌细胞开辟迁移通道
- I型胶原凝胶共培养实验显示:CAFs率先浸润后,癌细胞(尤其是肿瘤干细胞)迁移能力显著增强
- ECM降解降低组织硬度,这种机械阻力减少进一步促进癌细胞迁移
CAF靶向治疗策略
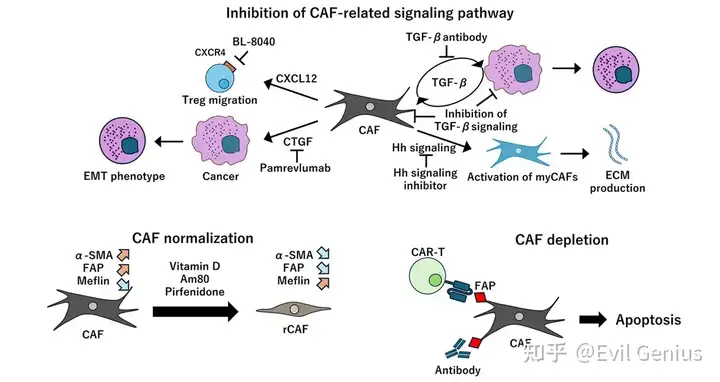
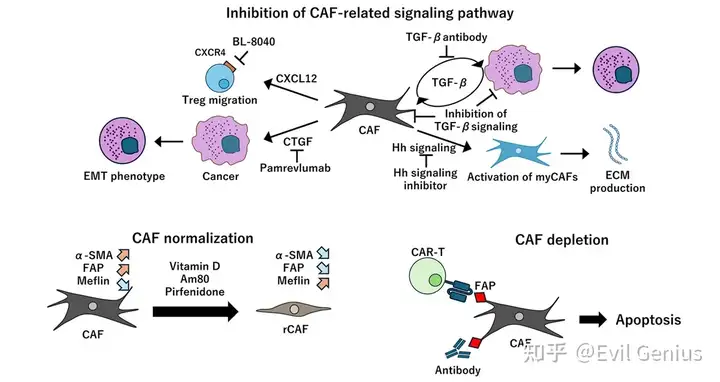
靶向CAF相关信号通路的治疗策略
在TME中,癌细胞通过分泌细胞因子驱使CAFs促肿瘤进展,阻断这一过程可能抑制肿瘤生长。
1. TGF-β通路抑制剂
- 科学依据:TGF-β不仅激活myCAFs分化,还由CAFs分泌诱导癌细胞EMT
- 临床现状: ✓ 过去十年开展多项TGF-β抑制剂联合免疫检查点抑制剂的临床试验 ✓ 动物模型显示抑制TGF-β可能加速胰腺癌进展 ✓ 临床试验疗效未达预期且出现副作用 ✓ 癌症EMT后抑制TGF-β可能适得其反,提示需根据癌症分期精准把握干预时机
2. CTGF靶向治疗
- 机制:CAFs分泌的CTGF通过调控增殖和EMT促进肿瘤进展,其高表达与不良预后相关
- 突破性进展:抗CTGF-1抗体pamrevlumab联合吉西他滨/白蛋白紫杉醇治疗胰腺癌时,显著提升疗效且未增加毒性
3. CXCR4-CXCL12轴抑制剂
- 作用机制:FAP+ CAFs激活CXCR4-CXCL12通路促进Tregs迁移(与乳腺癌不良预后相关)
- 临床证据:CXCR4拮抗剂BL-8040在PDAC患者中增加CD8+ T细胞浸润并减少Tregs
4. Hedgehog(Hh)信号通路调控
悖论现象:
✓ PDAC中癌细胞过表达的Shh(Hh家族配体)诱导CAFs富集的TME,但Shh缺失反而导致侵袭性肿瘤 ✓ 使用Smoothened拮抗剂抑制Hh信号: ▪ 减少抑癌型myCAFs,增加促癌型iCAFs ▪ 导致CD8+ T细胞减少和Tregs增加 ✓ 但Hh抑制可促进血管正常化,通过减少ECM增强吉西他滨渗透
- 临床结果:吉西他滨联合Hh抑制剂未能改善无进展生存期
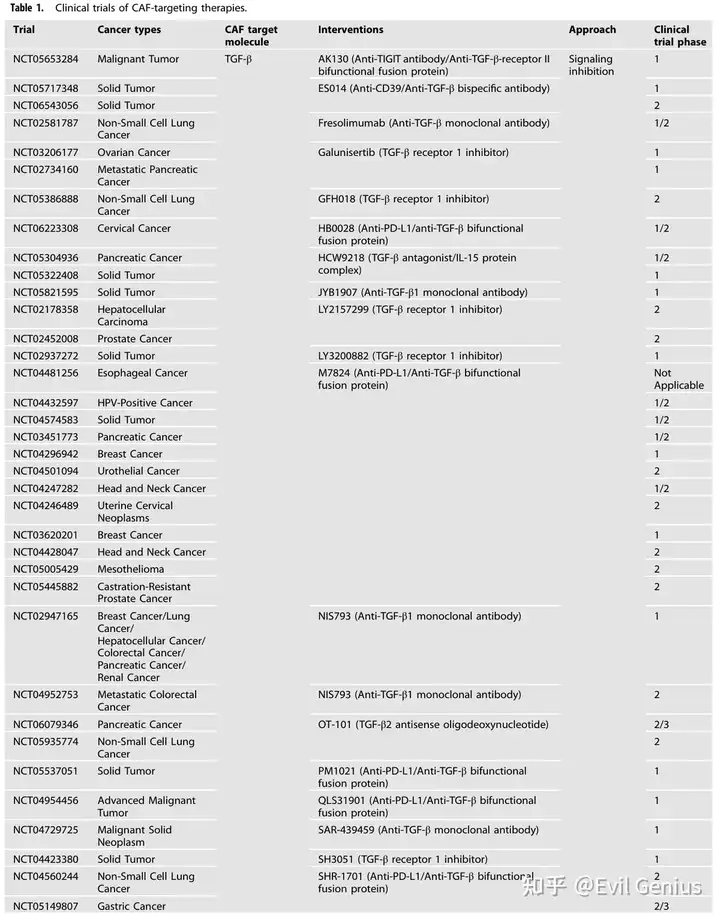
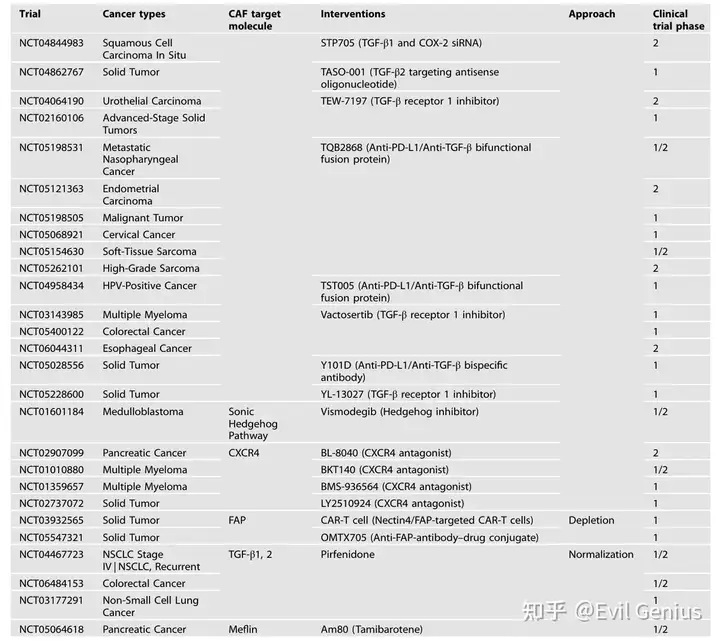
临床转化挑战
尽管CAF清除策略(尤其是靶向FAP+ CAFs的CAR-T疗法)在临床前研究中展现出:
- 降低胶原含量
- 改善T细胞浸润
但面临核心困境:
- 功能异质性:促癌与抑癌CAF亚型缺乏明确分子标记物
- 双刃剑效应:如PDAC模型中肌成纤维细胞清除反而加速肿瘤进展
- 选择困境:目前难以实现仅清除促瘤CAF亚型而保留抑瘤亚型
CAF功能正常化治疗策略
与正常组织中仅在伤口愈合时激活的成纤维细胞不同,肿瘤中的CAFs处于持续活化状态。因此,逆转CAFs的持续活化状态、减少ECM过度产生成为改善TME的新思路。
CAF研究未来方向
- 空间组学整合:结合单细胞与空间转录组解析CAFs三维分布及细胞互作网络
- 预后标志物开发:基于CAFs特征建立疗效预测体系
- 个体化治疗:患者特异性CAFs图谱指导精准医疗
- 联合治疗优化:CAF靶向与传统疗法联用克服耐药
CAFs至少包含五种功能异质的亚型,通过ECM过度产生、免疫抑制和血管生成等机制介导治疗抵抗。近年来研究不仅揭示了CAFs与其它细胞的复杂互作网络,更为突破性疗法的开发奠定基础。未来CAFs研究将继续推动涵盖TME调控的癌症治疗新范式发展。
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号