分析梳理--分子动力学模拟的常规步骤十(Gromacs)
原创
分析梳理--分子动力学模拟的常规步骤十(Gromacs)
原创
追风少年i
发布于 2026-05-19 10:03:14
发布于 2026-05-19 10:03:14
作者,Evil Genius
前面我们完成了动力学模拟,做了一些基础的分析,但是在数据分析的过程,需要我们分析更多的情况。
接下来我们就要做一些之前动力学模拟之后的相互作用分析。
首先:提取轨迹
gmx trjconv -f md_0_1_noPBC.xtc -s md_0_1.tpr -o 5.pdb -sep-b 49890 -e 49890 -n index.ndx-f:输入轨迹文件(如.xtc文件),并且为处理周期性之后的轨迹文件最佳
-s:输入模拟使用的.tpr文件
-sep:将每个帧写入单独的.gro、.g96或.pdb文件
-b:从轨迹读取第一帧的时间(默认单位ps)
-e:以轨迹读取最后一帧的时间(默认单位ps)
-n:索引文件
-o:输出文件
注:当-e和-b为一样的数值时,输出为某帧的结构
第二步:RMSF
gmx rmsf -s md_0_1.tpr -f md1.xtc -o rmsf.xvg -resgmx rmsf计算(可选地)拟合到参考坐标系(用-s表示)后轨迹中原子位置(用-f表示)的均方根波动(rmsf,即标准偏差)。可以表征蛋白质氨基酸在整个模拟过程中的柔性和运动剧烈程度。
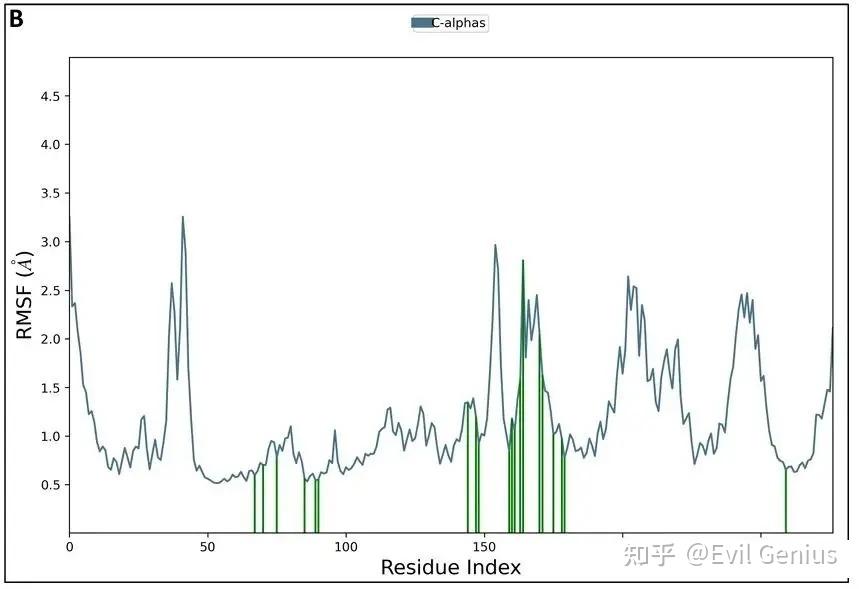
100 ns分子动力学模拟中蛋白的RMSF图
图表形式:横坐标为氨基酸残基序号(按序列顺序),纵坐标为每个残基的RMSF值(Å)。图中可能用绿色竖线标记与配体有接触的残基。
含义:RMSF (Root Mean Square Fluctuation, 根均方波动) 测量单个残基在模拟过程中的柔性/波动程度。与 RMSD 测量整体偏移不同,RMSF 关注每个氨基酸的“活跃度”。
研究可以借助RMSF探索蛋白质氨基酸的柔性,以及蛋白质柔性与配体结合活性氨基酸的关系。
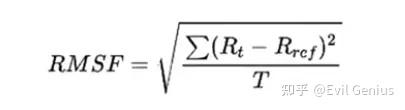
看一下参数
-f:输入轨迹文件(如.xtc文件),并且为处理周期性之后的轨迹文件最佳
-s:输入模拟使用的.tpr文件
-o:输出文件,一般为文本可读的xvg文件
-res:计算每个残基的平均值,这样输出文件为每个残基对应的RMSF值
分析的意义
识别柔性区域:峰值通常出现在loop区、末端(N端/C端),这是正常现象。α螺旋和β折叠区域的RMSF一般较低。
活性位点特征:与配体结合的关键残基通常表现为低RMSF(刚性),因为结合限制了其运动。若活性位点残基RMSF异常高,可能提示结合不稳定或模拟设置问题。
突变影响评估:比较野生型与突变体的RMSF图,若突变导致活性位点附近RMSF显著升高,可能削弱结合亲和力。
绿色竖条:图中标记的绿色竖条表示与配体有接触的残基。观察这些残基的RMSF值是否普遍较低,可验证结合界面的稳定性。
扩展(经过Gromacs处理后的蛋白质失去了b-factor信息,需要单独处理)。
gmx rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -ormsf_temp.xvg -ox rmsf_avg.pdb -res -oq rmsf_bfac.pdb命令可以利用RMSF模块输出含有B-factor信息的pdb文件,之后通过pymol可视化rmsf_bfac.pdb,通过pymol的b-factor来进行上色即可。
第三:配体径向分布
gmx rdf -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rdf_1_surf.xvg-n index.ndx -ref protein -sel 1ZIN -bin 0.01 -surf mol -b 85000-e 100000 -pbc yes径向分布函数可以表征两种类型的分子之间的距离关系。分析配体到蛋白质表面的径向分布函数,可以获得稳定时期配体到蛋白质表面的距离等信息。
GROMACS中A类型粒子和B类型粒子之间的径向分布函数(RDF)被定义为:
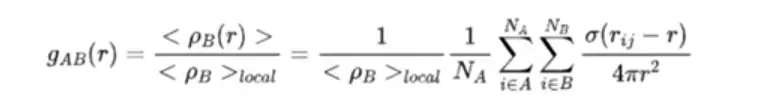
其中<p(r)>表示距离A粒子r距离,厚度为bin的壳层内B粒子的密度,p表示以A粒子为中心,半径的球内B粒子的平均密度。
通常径向分布函数用于分析连续相(比如说模拟体系中的水分子在蛋白质周围的径向分布),但是也可以利用其对蛋白质周围配体的存在进行分析。根据上述公式可得,将其应用到配体分析时,函数会呈现出与连续相的径向分布函数不一样的特征。对于半径为r的球体,配体的平均密度是一个固定值,但在某些壳层内,因为没有配体分布,故而会使得函数值为零,而在某些配体密集分布的地方,会使得函数值较高;而对于连续相,则函数最终应该会收敛到1附近(也即在半径很大的时候,壳层平均密度与总体平均密度接近),且函数峰值不会太高。
第四:氢键分析
gmx hbond -s md_0_1.tpr -f md_0_1_noPBC.xtc -num hnum.xvggmx hbond计算和分析氢键。氢键是根据氢-供体-受体的角度(零被延伸)和供体-受体的距离(或氢-受体使用-noda)的截止值来确定的。OH和NH基团被认为是供体,O总是受体,N默认是受体。假定虚拟氢原子与前面的第一个非氢原子相连。需要指定两组(如蛋白和小分子)进行分析,这两组必须相同或不重叠。分析了两个基团之间的所有氢键。
hbond程序使用几何准则来对氢键加以判定,当氢键供受体距离小于3.5埃且氢-供体-受体所成角度小于30度时,即认为其为一个氢键。
增加了-ac参数可以自动计算氢键的平均存在周期(forward lifetime),此参数可以作为衡量氢键稳定性的一个指标。
参数
-f:输入轨迹文件(如.xtc文件),并且为处理周期性之后的轨迹文件最佳
-s:输入模拟使用的.tpr文件
-n:索引文件(如果有的话)。
分析结果
-num:氢键数随时间的函数。
-ac:所有氢键的存在函数(0或1)的所有自相关的平均值。
dist:所有氢键的距离分布。
ang:所有氢键的角度分布。
-hx:n-n+i个氢键的个数与时间的关系,其中n和ni为残基数,i的取值范围为0~6。这包括蛋白质中与螺旋相关的n-n+3、n-n+4和n-n+5氢键。
-hbn:所有选定基团、选定基团的给体、氢原子和受体、所有基团的所有氢键原子和所有参与插入的溶剂原子。除非设置了-nomerge,否则输出是有限的。
-hbm:所有框架上所有氢键的存在矩阵,这也包含溶剂插入到氢键中的信息。排序与-hbn索引文件中的顺序相同。写出每个时间段内分析的供体和受体的数量。这在使用-shell时特别有用。
-nhbdist:计算每个氢的氢键数,以便将结果与拉曼光谱进行比较。
第五:自由能地貌
自由能形貌图(FEL,freeenergy landscape)可以表征物质在模拟过程中经历的自由能的变化。通过计算蛋白质和配体复合物的自由能形貌图可以给抽提复合物的特征构象提供指导。同时蛋白质在模拟的稳定时期的自由能形貌图可以表征蛋白质构象的稳定性。
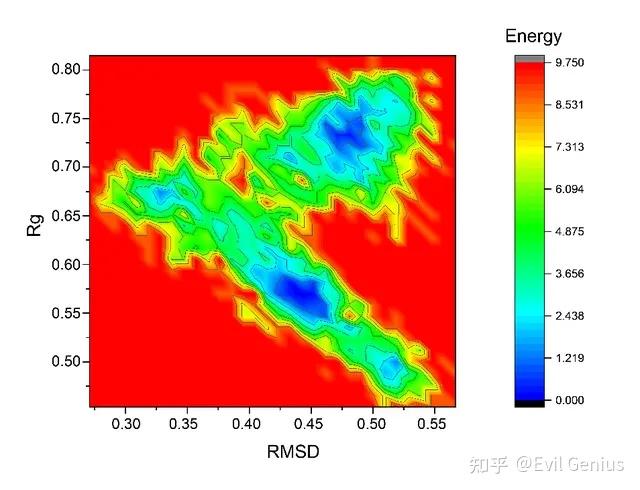
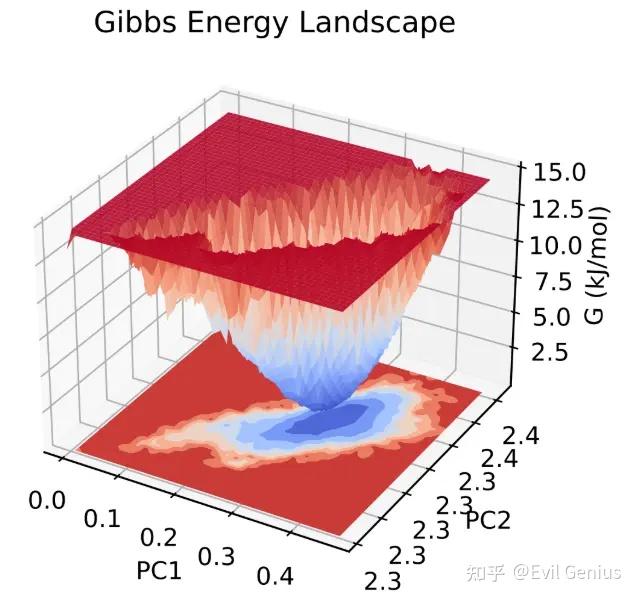
FEL在GROMACS中利用两个变量来进行表示,通常是分子的回旋半径和均方根误差(文献中常见的做法应当是首先做「主成分分析」,然后利用前两个主成分作为FEL的变量),GROMACS可以利用这两个变量来计算相应的自由能;在得到回旋半径、均方根误差、自由能三个维度的数据之后,即可以作图得到自由能形貌图。GROMACS得到的自由能通常不是准确真实的自由能数据,而是基于构象的分布来进行的Gibbs自由能估计,因此充分的模拟采样是十分必要的。
构建自由能形貌图的过程如下:
*抽提蛋白质配体复合物、蛋白质的RMSD
*抽提蛋白质配体复合物、蛋白质的回旋半径
*将回旋半径和RMSD的数据文件进行组合,第一列为时间,第二列第三列为相应时间下的回旋半径和RMSD数据
*设定温度为310K(模拟的温度),利用gmx sham命令获得自由能的xpm数据文件
*通过脚本将xpm转换成其他格式的文件并绘图
使用到的命令
gmx gyrate s prolig.tpr -f prolig_fit.xtc -o FEL_gyrate.xvg
gmx rms -s prolig.tpr -f prolig_fit.xtc -o FEL_rmsd.xvg
#组合回旋半径和RMSD数据,生成sham.xvg
gmx sham -f sham.xvg -Is FEL_sham.xpm -b 8500 -tsham 310 -nlevels 100
#利用的xpm2txt.py脚本将xpm转换为txt
python2 xpm2txt.py -f FEL_sham.xpm -o FEL_sham.txt
#自己写个脚本把FEL_sham.txt作图或者用origin等软件就可以了
#或者xpm2png.py脚本直接转化为图片
xpm2png.py -ip yes -f gibbs.xpm看一下核心参数
-tsham:设定温度-nlevels:设定FEL的层次数量f:读入组合文件-Is:输出自由能形貌图(Gibbs自由能)
-g:输出log文件
-Ish:焓的形貌图
-Iss:熵的形貌图
在上面的命令执行完成之后,会输出好几个文件,除了上文提到的,还包括一个index文件(bindex.ndx)和一个ener.xvg文件。我们最关心的是Gibbs自由能的形貌图,也即gibbs.xpm。
第六:MD轨迹可视化
MD体系中蛋白质和配体的运动轨迹可以直观地反映蛋白质和配体的运动以及结合情况,因而蛋白质和配体运动轨迹的可视化就尤为重要。
可以利用前述预平衡轨迹可视化的方法进行轨迹可视化,但是为了更方便地研究分子运动轨迹,将轨迹导出为pdb文件应该是更佳的方式。
使用如下命令从调整过周期性的模拟轨迹中导出pdb文件:
gmx trjconv -s md_0_1.tpr -f md_0_1_noPBC.xtc -otraj.pdb -dt 100上述语句从已经校正过周期性边界的轨迹文件中每隔100ps导出一帧到pdb文件,加入了蛋白质和配体的index文件可以只导出体系中的蛋白质和配体而忽略其它物质。
利用如下命令对载入pymol的pdb文件进行处理:
intra_fit prolig #将轨迹中的帧对齐A
dss state=1#计算二级结构并应用到所有帧
setall_states=1 #叠合所有帧(optional)
viewport640,480#设置画框
set ray_trace_frames, 1 #设置光线追踪
之后将每一帧导出为图片并利用ffmpeg合并即可
有的时候蛋白质的热运动有一些噪声,可以利用filter消除这些高频运动,只保留整体的运动。
gmx filter -s md_0_1.tpr -f md_0_1_noPBC.xtc -ol traj.pdb -dt100 -n prolig.ndx -fit -nf 5gmx filter用于对轨迹进行频率滤波.滤波器的形状为从-A到+A的cos(πt/A)+1,其中A为选项-nf与输入文件中时间步的乘积.对低通滤波,滤波器可将周期为A的涨落降低85%,周期为2A的降低50%,周期为3A的降低17%.程序可输出低通和高通滤波后的轨迹.
选项-ol输出低通滤波后的轨迹,每-nf输入帧输出一次.滤波器长度与输出间隔的比值保证了可很好地抑制高频运动的混淆,这非常有利于制作平滑的电影.此外,对与坐标有线性关系的性质,其平均值会保持不变,因为所有输入帧在输出中的权重都是相同的.当需要所有帧时,可使用-all选项.
选项-oh输出高通滤波后的轨迹.高通滤波后的坐标会加到结构文件中的坐标上.当使用高通滤波时,请使用-fit选项或保证所用轨迹已经叠合到结构文件中的坐标.
看一下核心参数
-f[<.xtc/.trr/...>] traj.xtc 输入 轨迹:xtc trr cpt trjgro g96 pdb tng
-s[<.tpr/.tpb/..>]topol.tpr 输入,可选结构+质量(db):tpr tpb tpa gro g96 pdb brk ent
-n [<.ndx>]index.ndx 输入,可选索引文件输出,
-ol[<.xtc/.trr/...>]lowpass.xtc,可选轨迹:xtc trr trj gro g96 pdb tng
-oh [<.xtc/.trr/...>]highpass.xtc;输出,可选轨迹:xtc trr trj gro g96 pdb tng
-nice 19 设置优先级
-b 0 从轨迹文件中读取的第一帧(ps)
-e 0 从轨迹文件中读取的最后一帧(ps)
-dt 0 只使用t除以dt的余数等于第一帧时间(ps)的帧,即两帧之间的时间间隔
-[no]w no查看输出的.xvg,.xpm,.eps和.pdb文件
-nf<int> 10 设置低通滤波的滤波器长度以及输出间距
-[no]all no输出所有低通滤波后的帧
-[no]nojump yes移除穿越盒子的原子跳跃
-[no]fit no将所有帧叠合到参考结构
将轨迹可视化之后,即可研究蛋白质和配体的相对运动,查看配体的结合位置、蛋白质的构象变化等等。
偶尔经过了周期性校正的轨迹导出的pdb文件在pymol无法正常cartoon显示,可以考虑换个周期性校正方式,比如说只保证分子完整,然后在pymol中对齐。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号