分子动力学--结合自由能的计算
原创
分子动力学--结合自由能的计算
原创
追风少年i
发布于 2026-05-24 09:00:18
发布于 2026-05-24 09:00:18
作者,Evil Genius
这一篇我们继续分子动力学。
以前高中不知道学的那些东西有什么用,现在多多少少知道一些了,生物这个学科,必须交叉,生物物理、生物化学、计算生物学、生物信息学等,才是正确的出路。
自由能是指在某一个热力学过程中,系统减少的内能中可以转化为对外做功的部分,它衡量的是:在一个特定的热力学过程中,系统可对外输出的“有用能量”。
自由能是物理化学上讲的,首先要明确一点,自由能是自发过程的判据:一个过程的自由能降低就能自发进行;能自发进行的过程,其自由能必然降低。
结合自由能变化=结合能(内能)变化+熵能变化例如:ΔA=ΔU-TΔS
备注:-TΔS,分子结合后,熵降低,即混乱程度降低,对应的熵能增加,所以公式里TΔS是负的。结合能ΔU是结合自由能ΔA的一部分,另一部分是熵能TΔS变化。
上述内容考察的是一个等温的过程,更加具体的的过程可以分为等温等容的过程和等温等压的过程。
等温等容过程中,ΔA称为亥姆霍兹自由能(Helmholzfreeenergy),亦称亥姆霍兹函数,又称为功函(workfunction),它显然是体系的状态函数。
在等温等容过程中,在无其他功时,亥姆霍兹自由能可逆的过程中保持不变,在不可逆的总是减少,直到最小时体系达到平衡。
等温等压过程中,ΔG称为吉布斯自由能(Gibbsfreeenergy)。体积要变化,多出一项对外做功PΔV。
ΔG= AH-TΔS (H=U+PV)
在等温过程等压中,在无其他功时,Gibbs自由能可逆的过程中保持不变,在不可逆的总是减少,直到最小时体系达到平衡。
自由能分类
具体是什么自由能取决于你模拟使用什么系综,NVT还是NPT。G=A+PV(P为压力,V为体积)。在生物的动力学反应中,因为Δ(PV)可以忽略不计,所以两者是相同的。
计算结合自由能的意义
反映结合亲和力,用于评价结合强度。结合自由能越负值,表明结合亲和力越强。
预测结合平衡常数,进而分析结合对平衡的影响。
比较不同配体与受体的相对结合能力,为药物设计提供指导。
分析关键残基对结合贡献,指导突变实验设计。
计算溶剂化自由能变化,了解溶剂效应对结合的影响。
计算配体结构变化对结合自由能的影响,为优化结构提供参考。
常用来做结合自由能计算的就是MM/PB(GB)SA。
该方法全称分子力学/泊松-波尔兹曼(广义波恩)表 面 积 (Molecular Mechanics / Poisson Boltzmann(Generalized Born) Surface Area)。顾名思义,该方法将结合自由能拆分成分子力学项和溶剂化能分别计算。
基本原理
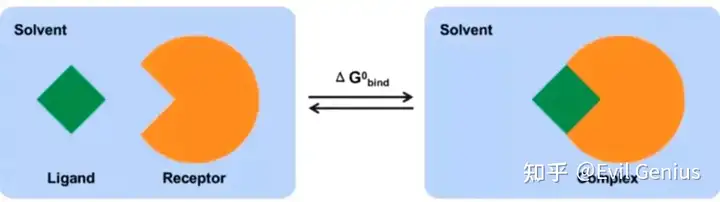
计算两个溶剂化分子在结合(bound)和游离(unbound)状态的结合自由能之差或者比较同一个分子不同的溶剂化构象的自由能。
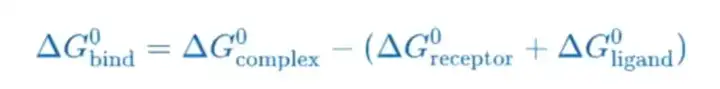
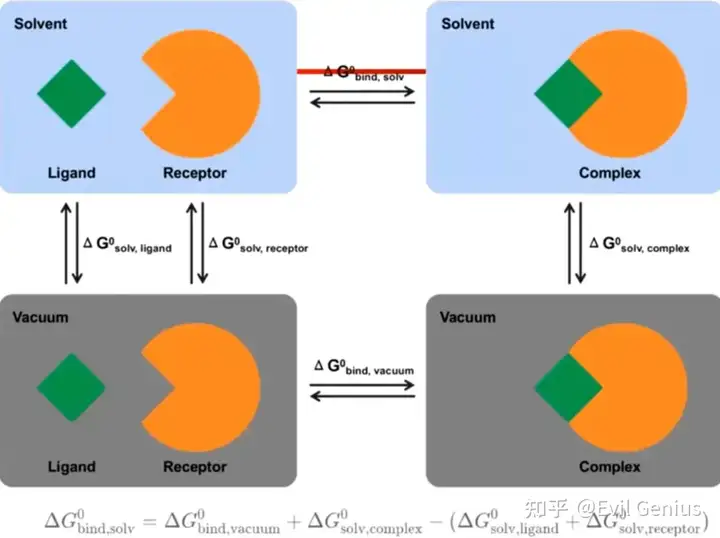
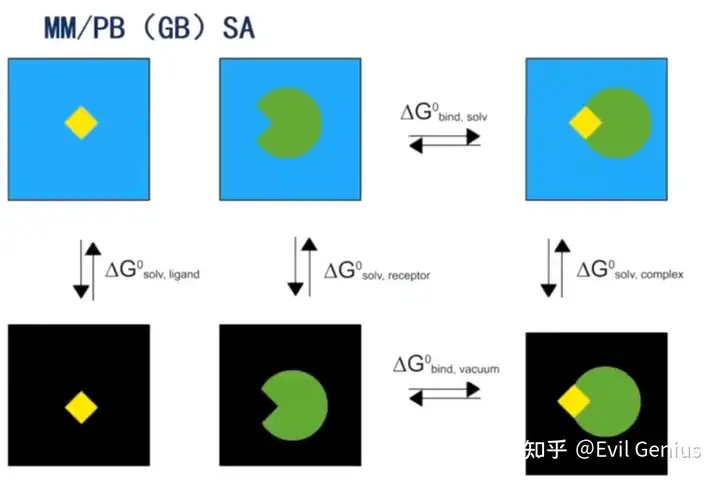
将溶剂中的总结合自由能分拆成分子力学项(真空中的结合自由能)和溶剂化能两部分分别计算.
分子力学项
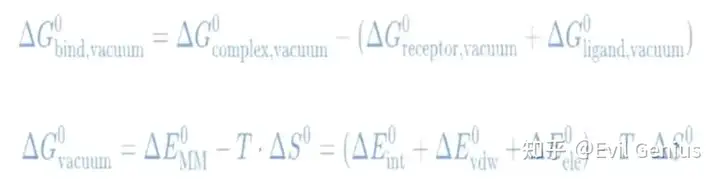
溶剂化能
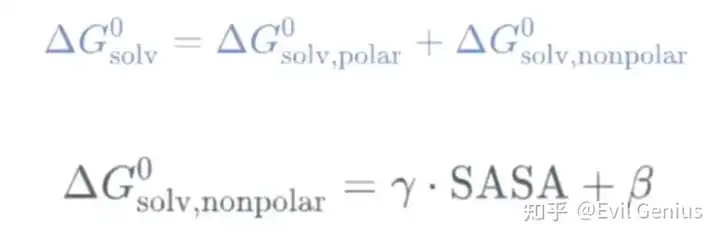
最后进行合并

PB VS GB
静电力学项计算不同:PBSA通过Poisson-Boltzmann方程;GBSA通过Born模型近似。
PBSA计算耗时,但考虑溶剂化和离子效应;GBSA计算快,但近似处理溶剂化。
PBSA更适合处理生物分子间相互作用;GBSA也可用于小分子和药物分子。
PBSA可提供绝对结合自由能值;GBSA通常给相对结合自由能。PBSA可考虑多组分溶解环境;GBSA多应用于单溶剂水溶液。
总体而言,PBSA理论上更准确全面,但计算量大;GBSA计算更高效,但存在一定近似。两者互补应用可获得生物分子结合自由能信息。
输入文件
1.生成tpr文件(md_0_1.tpr),预平衡;
2.运行成品模拟,得到xtc轨迹文件(md_0_1.xtc):
3.生成ndx文件(index.ndx),蛋白(Protein),配体(MOL)。也可以代表任意分子,比如两个有机小分子,而不一定就是蛋白和配体;索引组只能存在一个该名称选项
4.处理轨迹(md_0_1_noPBC.xtc)。如果存在二聚体,团簇等情况,确保组成的原子间没有分离,即处理好体系的周期性。
计算参数
step=0#计算MMPBSA时从轨迹的第几步开始运行
gmx='gmx’#gromacs运行的路径以及程序,一般配置环境变量后只输入gmx即可
dump=“$gmx dump”#将二进制的tpr文件转化成文本文件,以便抽取所需的参数.采用这种方法,可以避免版本不兼容的问题,因此也就可以支持任意版本的GROMACS
trjconv=“$gmx trjconv -dt 1000”#用于处理轨迹的周期性叠合问题,尽量自己处理完成,特别是在需要对周期性进行特殊处理的情况下;使用-dt,可以减少输出中的帧数
converttpr=“$gmx convert -tpr"#生成计算适合的体系组分的tpr文件
apbs=/data1_nvme/APBS-3.0.0.Linux/bin/apbs' #apbs程 序 的绝对路径
PBSA部分的计算是借助APBS程序完成,因此,在使用gmx_mmpbsa前,必需安装好APBS程序
了解静电相互作用对于研究生物分子过程是必不可少的。通过结构基因组学和其他努力,蛋白质和其他生物聚合物的结构正以越来越快的速度被确定。为了将这些信息整合到药物发现或其他应用的物理模型中,需要有能力评估生物聚合物内部和之间的能量相互作用。在分子能量学的各种组分中,溶剂化性质和静电相互作用特别重要,因为这些相互作用的范围很长,而且典型的生物聚合物组分具有大量的电荷。APBS解决了大型生物分子组合的连续统静电方程。
主要运行过程
其他参数主要是APBS计算所用的极性参数,非极性参数,网格参数,一般无需更改太多
脚本首先处理轨迹:1.完整化;2.居中叠合.之后将构型输出到pdb文件。
脚本其次抽取tpr中的原子信息,存放在qrv文件中。主要是复合物中每个原子的电荷,半径,L参数以及残基信息。
脚本根据pdb文件中原子的坐标获取APBS网格参数,并将每帧构型输出到APBS所需的pqr文件,同时生成APBS输入文件*.apbs.然后调用APBS计算每帧构型对应的apbs文件,并计算极性PB,非极性SA部分的贡献,再计算MM贡献,同时进行残基分解,输出结果。


生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号