
springboot第14集:MyBatis-CRUD讲解

注意点:增、删、改操作需要提交事务!
- 为了规范操作,在SQL的配置文件中,我们尽量将Parameter参数和resultType都写上!
编写Mapper接口类
import com.da.pojo.User;
import java.util.List;
public interface UserMapper {
List<User> selectUser();
}编写Mapper.xml配置文件
<?xml version="1.0" encoding="UTF-8" ?>
<!DOCTYPE mapper
PUBLIC "-//mybatis.org//DTD Mapper 3.0//EN"
"http://mybatis.org/dtd/mybatis-3-mapper.dtd">
<mapper namespace="com.da.dao.UserMapper">
<select id="selectUser" resultType="com.da.pojo.User">
select * from user
</select>
</mapper>select
- select标签是mybatis中最常用的标签之一
- select语句有很多属性可以详细配置每一条SQL语句
- SQL语句返回值类型。
- 传入SQL语句的参数类型 。
- 命名空间中唯一的标识符
- 接口中的方法名与映射文件中的SQL语句ID 一一对应
- id
- parameterType
- resultType
在UserMapper中添加对应方法
public interface UserMapper {
//查询全部用户
List<User> selectUser();
//根据id查询用户
User selectUserById(int id);
}在UserMapper.xml中添加Select语句
<select id="selectUserById" resultType="com.da.pojo.User">
select * from user where id = #{id}
</select>在UserMapper接口中添加对应的方法
//添加一个用户
int addUser(User user);在UserMapper.xml中添加insert语句
<insert id="addUser" parameterType="com.da.pojo.User">
insert into user (id,name,pwd) values (#{id},#{name},#{pwd})
</insert>//修改一个用户
int updateUser(User user);<update id="updateUser" parameterType="com.da.pojo.User">
update user set name=#{name},pwd=#{pwd} where id = #{id}
</update>根据id删除一个用户
//根据id删除用户
int deleteUser(int id);编写对应的配置文件SQL
<delete id="deleteUser" parameterType="int">
delete from user where id = #{id}
</delete>select * from foo where bar like #{value}
select * from foo where bar like "%"#{value}"%"configuration(配置)
properties(属性)
settings(设置)
typeAliases(类型别名)
typeHandlers(类型处理器)
objectFactory(对象工厂)
plugins(插件)
environments(环境配置)
environment(环境变量)
transactionManager(事务管理器)
dataSource(数据源)
databaseIdProvider(数据库厂商标识)
mappers(映射器)
<!-- 注意元素节点的顺序!顺序不对会报错 --><environments default="development">
<environment id="development">
<transactionManager type="JDBC">
<property name="..." value="..."/>
</transactionManager>
<dataSource type="POOLED">
<property name="driver" value="${driver}"/>
<property name="url" value="${url}"/>
<property name="username" value="${username}"/>
<property name="password" value="${password}"/>
</dataSource>
</environment>
</environments>子元素节点:environment
- 具体的一套环境,通过设置id进行区别,id保证唯一!
子元素节点:transactionManager - [ 事务管理器 ]
<!-- 语法 -->
<transactionManager type="[ JDBC | MANAGED ]"/>子元素节点:数据源(dataSource)
- dataSource 元素使用标准的 JDBC 数据源接口来配置 JDBC 连接对象的资源。
- 数据源是必须配置的。
type="[UNPOOLED|POOLED|JNDI]")- unpooled:这个数据源的实现只是每次被请求时打开和关闭连接。
- pooled:这种数据源的实现利用“池”的概念将 JDBC 连接对象组织起来 , 这是一种使得并发 Web 应用快速响应请求的流行处理方式。
- jndi:这个数据源的实现是为了能在如 Spring 或应用服务器这类容器中使用,容器可以集中或在外部配置数据源,然后放置一个 JNDI 上下文的引用。
- 数据源也有很多第三方的实现,比如dbcp,c3p0,druid等等….
Mapper文件
<?xml version="1.0" encoding="UTF-8" ?>
<!DOCTYPE mapper
PUBLIC "-//mybatis.org//DTD Mapper 3.0//EN"
"http://mybatis.org/dtd/mybatis-3-mapper.dtd">
<mapper namespace="com.da.mapper.UserMapper">
</mapper>- namespace中文意思:命名空间,作用如下:
- namespace的命名必须跟某个接口同名
- 接口中的方法与映射文件中sql语句id应该一一对应
- namespace和子元素的id联合保证唯一 , 区别不同的mapper
- 绑定DAO接口
- namespace命名规则 : 包名+类名
MyBatis 的真正强大在于它的映射语句,这是它的魔力所在。由于它的异常强大,映射器的 XML 文件就显得相对简单。如果拿它跟具有相同功能的 JDBC 代码进行对比,会立即发现省掉了将近 95% 的代码。MyBatis 为聚焦于 SQL 而构建,以尽可能地减少麻烦。
db.properties
driver=com.mysql.jdbc.Driver
url=jdbc:mysql://localhost:3306/mybatis?useSSL=true&useUnicode=true&characterEncoding=utf8
username=root
password=123456typeAliases优化
`<!--配置别名,注意顺序-->`
`<typeAliases>`
`<typeAlias type="com.da.pojo.User" alias="User"/>`
`</typeAliases>`无论是 MyBatis 在预处理语句(PreparedStatement)中设置一个参数时,还是从结果集中取出一个值时, 都会用类型处理器将获取的值以合适的方式转换成 Java 类型。
可以重写类型处理器或创建自己的类型处理器来处理不支持的或非标准的类型。
MyBatis 每次创建结果对象的新实例时,它都会使用一个对象工厂(ObjectFactory)实例来完成。默认的对象工厂需要做的仅仅是实例化目标类,要么通过默认构造方法,要么在参数映射存在的时候通过有参构造方法来实例化。如果想覆盖对象工厂的默认行为,则可以通过创建自己的对象工厂来实现。
作用域理解
- SqlSessionFactoryBuilder 的作用在于创建 SqlSessionFactory,创建成功后,SqlSessionFactoryBuilder 就失去了作用,所以它只能存在于创建 SqlSessionFactory 的方法中,而不要让其长期存在。因此 SqlSessionFactoryBuilder 实例的最佳作用域是方法作用域(也就是局部方法变量)。
- SqlSessionFactory 可以被认为是一个数据库连接池,它的作用是创建 SqlSession 接口对象。因为 MyBatis 的本质就是 Java 对数据库的操作,所以 SqlSessionFactory 的生命周期存在于整个 MyBatis 的应用之中,所以一旦创建了 SqlSessionFactory,就要长期保存它,直至不再使用 MyBatis 应用,所以可以认为 SqlSessionFactory 的生命周期就等同于 MyBatis 的应用周期。
- 由于 SqlSessionFactory 是一个对数据库的连接池,所以它占据着数据库的连接资源。如果创建多个 SqlSessionFactory,那么就存在多个数据库连接池,这样不利于对数据库资源的控制,也会导致数据库连接资源被消耗光,出现系统宕机等情况,所以尽量避免发生这样的情况。
- 因此在一般的应用中我们往往希望 SqlSessionFactory 作为一个单例,让它在应用中被共享。所以说 SqlSessionFactory 的最佳作用域是应用作用域。
- 如果说 SqlSessionFactory 相当于数据库连接池,那么 SqlSession 就相当于一个数据库连接(Connection 对象),可以在一个事务里面执行多条 SQL,然后通过它的 commit、rollback 等方法,提交或者回滚事务。所以它应该存活在一个业务请求中,处理完整个请求后,应该关闭这条连接,让它归还给 SqlSessionFactory,否则数据库资源就很快被耗费精光,系统就会瘫痪,所以用 try…catch…finally… 语句来保证其正确关闭。
- 所以 SqlSession 的最佳的作用域是请求或方法作用域。
@PreAuthorize("@ss.hasPermi('system:good:list')")
@GetMapping("/list")
public TableDataInfo list()
{
startPage();
List<GoodsGood> list = goodsGoodService.selectGoodsGoodList();
return getDataTable(list);
}<el-col :span="1.5">
<el-button
type="warning"
plain
icon="el-icon-download"
size="mini"
@click="handleExport"
v-hasPermi="['goods:good:export']"
>导出</el-button>
</el-col>
handleExport() {
this.download('goods/good/export', {
...this.queryParams
}, `good_${new Date().getTime()}.xlsx`)
}
image.png
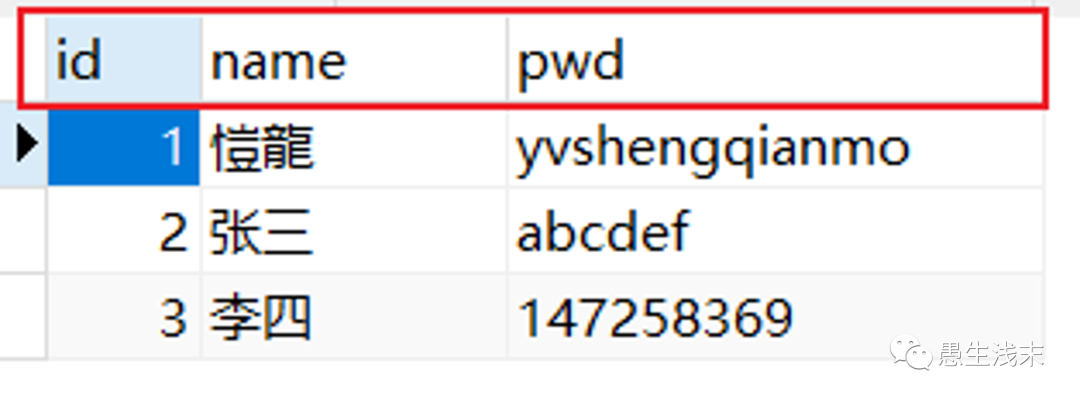
public class User {
private int id; //id
private String name; //姓名
private String password; //密码和数据库不一样!
//构造
//set/get
//toString()
}//根据id查询用户
User selectUserById(int id);<select id="selectUserById" resultType="user">
select * from user where id = #{id}
</select>
<select id="selectUserById" resultType="User">
select id , name , pwd as password from user where id = #{id}
</select>@Test
public void testSelectUserById() {
SqlSession session = MybatisUtils.getSession(); //获取SqlSession连接
UserMapper mapper = session.getMapper(UserMapper.class);
User user = mapper.selectUserById(1);
System.out.println(user);
session.close();
}使用结果集映射->ResultMap
<resultMap id="UserMap" type="User">
<!-- id为主键 -->
<id column="id" property="id"/>
<!-- column是数据库表的列名 , property是对应实体类的属性名 -->
<result column="name" property="name"/>
<result column="pwd" property="password"/>
</resultMap>
<select id="selectUserById" resultMap="UserMap">
select id , name , pwd from user where id = #{id}
</select>自动映射在 MyBatis 中扮演着非常重要且强大的角色,其中 resultMap 元素更是起到了关键作用。使用它可以让繁琐的 JDBC ResultSet 数据提取代码中解放出来,甚至对于连接复杂语句的映射代码,一份 resultMap 也能代替数千行的代码。
ResultMap 的设计思想非常巧妙,简单语句无需配置显式的结果映射,而对于稍微复杂一点的语句,只需要描述它们之间的关系即可。
<select id="selectUserById" resultType="map">
select id , name , pwd
from user
where id = #{id}
</select>将所有的列映射到 HashMap 的键上,这由 resultType 属性指定。
<select id="selectUserById" resultMap="UserMap">
select id , name , pwd from user where id = #{id}
</select>
<resultMap id="UserMap" type="User">
<!-- id为主键 -->
<id column="id" property="id"/>
<!-- column是数据库表的列名 , property是对应实体类的属性名 -->
<result column="name" property="name"/>
<result column="pwd" property="password"/>
</resultMap>仓库地址:https://github.com/webVueBlog/JavaGuideInterview









腾讯云开发者

