10X单细胞空间数据分析之富集篇
原创

一、单细胞基因富集分析算法
一个不算正式的引言:目前来说,基于基因集进行分析已经开发出来了很多成体系的R包或者流程,理解来看,基因集评分其实就是自定义一个评分,然后来衡量目标基因集在某组织的表达情况,进而来推断其功能富集情况,所以说,这个给了我们以提示,算法是一定的,但是参考基因集可以是不同的,比如说铁死亡、铜死亡、细胞衰老等等
目前来看,常见的针对单细胞的基因集富集分析算法有:GSEA、GSVA、AddModuleScore、AUCell、ssGSEA以及UCell等等,关于其中的鉴别以及或多或少的区别,如果只是单纯的跑流程可能并不需要我们掌握。
但是对于我们分析人员而言,进一步的学习可以帮助我们充分的理解算法本身,也可以在有众多选择中选择自己认为正确的,那么接下来简单的来聊一聊上述这些算法:
1.GSEA
GSEA的输入文件是一个表达矩阵,我们需要提前把样本进行分组,然后对所有基因基于分组来进行排序,那么这个时候我们最常使用的排序方式就是通过LogFC的大小来进行排序(从大到小),这种排序天生就具有了一种属性即可以表示两组间基因的表达量变化趋势,排序之后的基因列表其顶部可以看作是上调的差异基因,其底部可以看作是下调的差异基因
那么这个时候GSEA分析其实回答我们的就是: 我们所选择的参考基因集是富集在这个基因排序列表中的顶部还是底部,如果在顶部,那么很显然我们选择的参考基因集就是呈现上调趋势,反之则是下调趋势,一般来说我们选择的分组都是疾病组和对照组,那么有了趋势我们就可以回答疾病组表达的是参考基因集功能(炎症、衰老、铜死亡)是高表达还是低表达,这样其实就是很好的规避掉了单纯的ORA算法所带来的局限性
一、单细胞基因富集分析算法
一个不算正式的引言:目前来说,基于基因集进行分析已经开发出来了很多成体系的R包或者流程,理解来看,基因集评分其实就是自定义一个评分,然后来衡量目标基因集在某组织的表达情况,进而来推断其功能富集情况,所以说,这个给了我们以提示,算法是一定的,但是参考基因集可以是不同的,比如说铁死亡、铜死亡、细胞衰老等等
目前来看,常见的针对单细胞的基因集富集分析算法有:GSEA、GSVA、AddModuleScore、AUCell、ssGSEA以及UCell等等,关于其中的鉴别以及或多或少的区别,如果只是单纯的跑流程可能并不需要我们掌握。
但是对于我们分析人员而言,进一步的学习可以帮助我们充分的理解算法本身,也可以在有众多选择中选择自己认为正确的,那么接下来简单的来聊一聊上述这些算法:
1.GSEA
GSEA的输入文件是一个表达矩阵,我们需要提前把样本进行分组,然后对所有基因基于分组来进行排序,那么这个时候我们最常使用的排序方式就是通过LogFC的大小来进行排序(从大到小),这种排序天生就具有了一种属性即可以表示两组间基因的表达量变化趋势,排序之后的基因列表其顶部可以看作是上调的差异基因,其底部可以看作是下调的差异基因
那么这个时候GSEA分析其实回答我们的就是: 我们所选择的参考基因集是富集在这个基因排序列表中的顶部还是底部,如果在顶部,那么很显然我们选择的参考基因集就是呈现上调趋势,反之则是下调趋势,一般来说我们选择的分组都是疾病组和对照组,那么有了趋势我们就可以回答疾病组表达的是参考基因集功能(炎症、衰老、铜死亡)是高表达还是低表达,这样其实就是很好的规避掉了单纯的ORA算法所带来的局限性
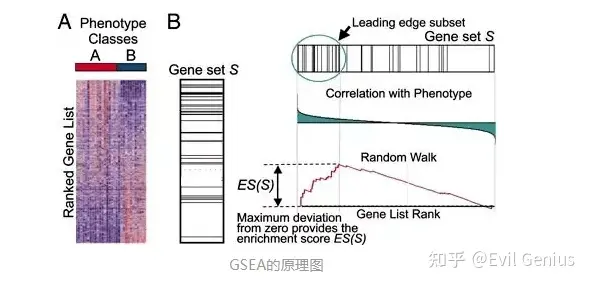
注意:
1.GSEA要求的输入数据是全部表达矩阵,并不是经过筛选后的表达矩阵
2.GSEA是一种算法,如果你是通过GO数据库进行GSEA富集分析,那么应该被叫做GO_GSEA富集分析,其实简单理解就是,我们提供参考基因集然后进行GSEA富集分析去探索我们的参考基因集在实验组究竟是上调还是下调
3.对于是否进行批次效应的去除,因为我们需要进行差异分析获得可以排序的依据,所以这里笔者认为我们需要进行批次效应的去除,因为需要进行差异分析获得LogFC,但是简单来说就是输入数据其实就是可以进行差异分析的输入数据即可
2.GSVA
GSVA富集分析全称叫做基因集变异分析,在这里说一下个人的简单理解,其实我们可以把其看作是一个特殊的富集分析,为什么说是特殊呢,GSVA的观测不再是Gene symbol而是转换成了通路名称,所以我们可以通过GSVA获得表达矩阵转换成通路表达矩阵后的结果,然后我们对转换后的结果进行差异分析,就可以得到通路LogFC的情况

注意:
1.GSVA选择不同的参考基因集得到的结果是不一样的,这一点我们可以灵活运用
2.输入数据可以是counts也可以是logTPM,如果是counts则参数kcdf需要选择"Poisson",如果是logTPM则参数kcdf需要选择"Gaussian"
3.AddModuleScore
关于AddModuleScore函数,我曾经写过系统的推文,感兴趣的小伙伴可以直接点击下方链接进行跳转阅读:
Seurat包的打分函数AddModuleScore
虽然可能理解算法流程对我们使用可能没有太多的帮助,但是这里依旧想要简单的概括一下算法的大概流程:通过计算参考基因集在表达矩阵中所有基因的平均表达量,然后通过平均表达量把表达矩阵切割成若干份,然后从切割的部分中随机抽取这些切块作为背景基因,知道这些的重点其实是下面这句话既然其中存在随机,那么即使使用相同参考基因集为相同数据进行打分,也可能会产生不同的结果
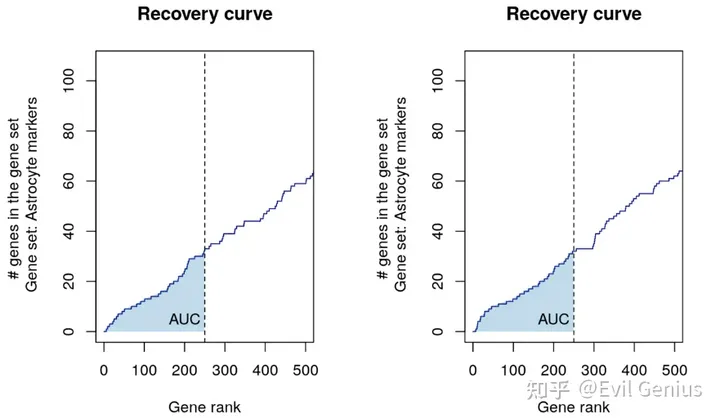
4.AUCell
首先我们通过尽量简单的语言来回答一下AUCell算法,其实本质上就是计算我们所选择的参考基因集是否在单细胞中每个变量上富集,因为单细胞数据的变量是细胞ID,我们也可以说是计算是否在细胞上富集
简单来说AUCell算法分为三个步骤:
1.针对每个细胞中的每个基因进行排序(既然涉及到了排序,那么显然标准化与否对于我们的影响就可以忽略不计了,因为即使标准化也不会改变谁大谁小这个直观的事实)
2.用过使用AUC来评估我们的参考基因集是否在每个细胞前5%表达基因集内富集,然后进而来评估参考基因集针对每个细胞的富集情况
5.ssGSEA
ssGSEA全名其实被称为"单样本基因集富集分析",从名字其实我们就可以看出,其是GSEA方法的扩展,既然是扩展,那么我们就首先需要知道GSEA算法的局限性
GSEA算法的局限其实就是我们需要提供排序信息,而我们的排序信息在很多情况下其实源自不同分组下差异表达,也就是logFC,那么这个时候问题来了:如果我只有一个分组,那么我如何评估某个基因集在单样本(一个分组)中的表达情况呢?
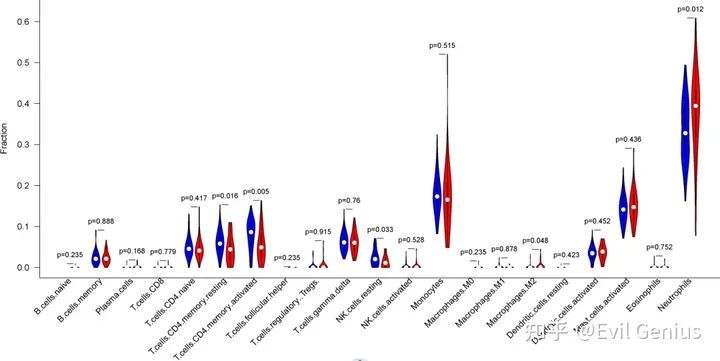
那么这个时候自然而然就诞生了ssGSEA,该算法可以让单个样本获得对应参考基因集的富集评分,通过这种形式,我们就可以把单一样本的基因表达谱转换为基因富集分数表达谱,那么这个时候我们就可以直观描述单一样本下针对参考基因集富集分数的高低,那么这个时候问题又来了:如果我有两个分组,我使用ssGSEA,可以吗?
那么这个时候我们可以参考免疫浸润分析答案自然就呼之欲出了,也就是说我如果把参考基因集换成免疫相关基因,然后通过使用ssGSEA针对疾病组和对照组,那么我们就可以得到疾病组和对照组分别的免疫浸润情况,然后通过进行秩和检验,自然就可以得到下面这张在文献中经常出现的小提琴图:
6.UCell
看到这里,相信我们都或多或少会有一种感觉,算法很多,但是都是得到一个打分,一个可以评估我们参考基因集在样本中表达情况的打分,那么很显然有了这层理解后,我们后面哪怕再接触其它"打分"算法,我们也可以知道我们的重心更应该放在参考基因集的选择上,也就是我们想要明确哪些功能在我们的样本中
那么回到算法本身,UCell算法具有以下特点:
1.对数据集本身要求不高,并不会受到数据集异质性的影响
2.对于处理大型数据具有一定优势
3.可以方便衔接Seurat包,快捷方便
好,那么介绍到这里,相信各位小伙伴依然还有疑惑,上述看似干货的东西显然并不能使我们掌握单细胞基因集富集分析,那么这里晨曦自然并不是仅仅止步于此
二、R包实践
接下来给大家介绍一个可以自用定制单细胞基因集富集分析需求同时可视化也很美观的R包——irGSEA包
接下来将直接演示代码,
library(Seurat)
library(SeuratData)
library(UCell)
library(irGSEA)
# loading datasetdata( "pbmc3k.final")
pbmc3k. final<- UpdateSeuratObject(pbmc3k. final) #
plotDimPlot(pbmc3k. final, reduction = "umap", group.by = "seurat_annotations",label = T) + NoLegend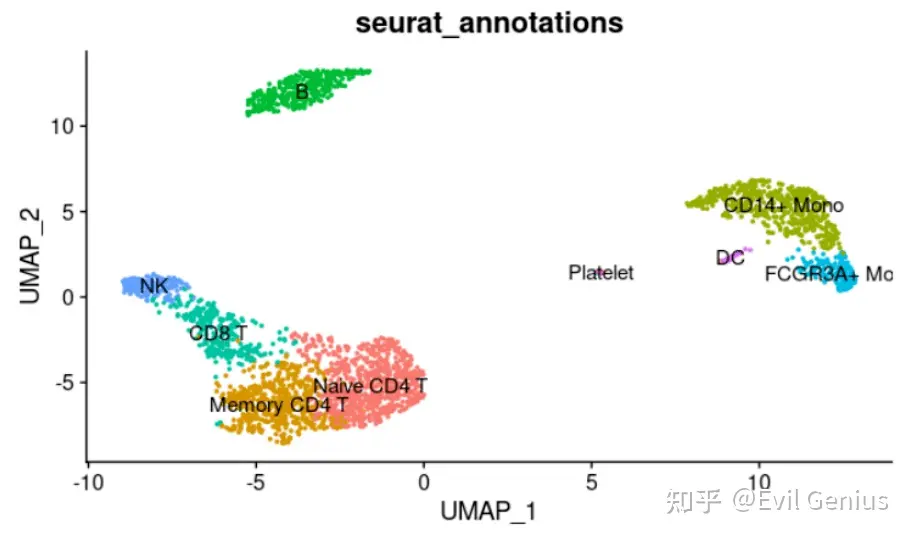
这个数据就是Seurat官网上经典的PBMC数据,我们这里演示的流程可以无缝对接自己的Seurat流程数据
Idents(pbmc3k. final) <- pbmc3k. final$seurat_annotations
#设置亚群,后续选择注释后的数据
#计算评分
pbmc3k. final<- irGSEA.score(object = pbmc3k. final, assay = "RNA", slot = "data", seeds = 123, ncores = 1, min.cells = 3, min.feature = 0, custom = F, geneset = NULL, msigdb = T, species = "Homo sapiens", category = "H", subcategory = NULL, geneid = "symbol", method = c( "AUCell", "UCell", "singscore", "ssgsea"), aucell.MaxRank = NULL, ucell.MaxRank = NULL, kcdf = 'Gaussian')
#整合差异基因集
result.dge <- irGSEA.integrate(object = pbmc3k. final, group.by = "seurat_annotations", metadata = NULL, col.name = NULL, method = c( "AUCell", "UCell", "singscore", "ssgsea"))一步函数即可完成相关分析,在这里作者内置了MSigDB数据库的相关基因集,当然R包本身也支持自定义基因集,这一块就给了我们无限的可能,其实就是更加的自由
#可视化
irGSEA.heatmap.plot < -irGSEA.heatmap( object= result.dge,method= "RRA", top= 50,show.geneset= NULL)
irGSEA.heatmap.plot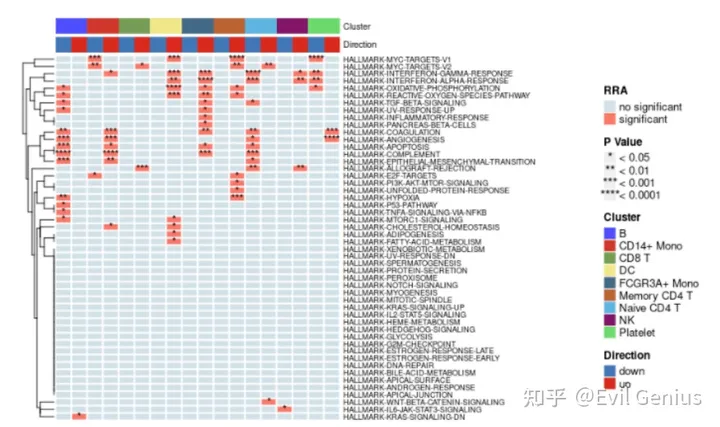
#可视化
irGSEA.barplot.plot <- irGSEA.barplot( object= result.dge, method = c( "AUCell", "UCell", "singscore", "ssgsea"))
irGSEA.barplot.plot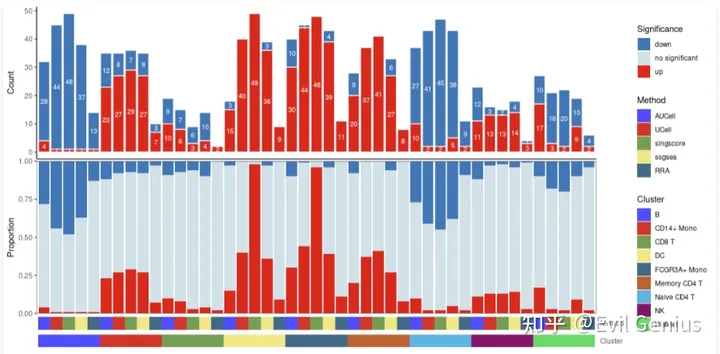
当然还有更多可视化的展示形式,大家感兴趣可以去作者的github上进行进一步的学习......
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号